


INTRODUCTION
Congenital cataract (CC) is defined as the occurrence of cataract within the first year of a child’s life.[1] The overall prevalence of CC ranges from 0.63 to 9.74 per 10,000 children.[2] It is one of the leading causes of visual disability in children, accounting for approximately 10% of childhood blindness around the world, which has brought significant burdens to their families as well as global socioeconomic status.[3] The etiology and clinical features of CC are multifaceted. The condition can manifest in one or both eyes, with variations in morphology, anatomical location, and severity of lens opacities. Most CC cases remain stable, while some may progress overtime.[4] Moreover, CC can occur in isolation or alongside other ocular or systemic developmental anomalies such as microphthalmia, microcornea, and/or iris anomalies.[5-6]
CC can be classified into different types based on the anatomical landmarks of the lens, cataract morphology, and etiology. However, there is a lack of a unified categorization method. Relying onone specific categorization systems in clinical practice, it is challenging for clinicians in making precise treatment strategies. Therefore, the objective of this review is to provide an overview of the status quo of CC categorization systems, including the categorization principle and applications, while highlighting areas in need of further study and future directions.
By far, there has been no unified standard for the categorization of CC. In the early stage, specific cataracts were described based upon the name of investigating doctor or the patients’ pedigree, such as Marner cataract[7] and Coppock cataract.[8] In 1942, Adamas first described Coppock cataract as a circular spotted disc in the center of the lens under slit-lamp examination.[9] Subsequently, scholars began to classify CC based on the morphology and anatomical locations of lens opacities.[10] Meanwhile, in pursuit of understanding the pathogenesis of cataracts and pursuing etiological treatment, the etiology of CC was also used as a classification criterion. With the advancement of medical technology, innovative approaches such as genome-wide arrays, whole genome sequencing technologies, and advanced ocular examinations had been employed to classify CC. Gene sequencing technology facilitated the exploration of the relationship between causative genes and cataracts, and scholars tried to classify CC based on the correlation between genotype and phenotype.[11] Recently, advancements in ultrasonic and optical measurement technology have enabled the identification of more detailed characteristics of the anterior segment by various measuring instruments.[12-14] Classification systems for CC based on the outcomes of anterior segment measuring instruments have emerged to investigate the association between CC and other anterior segment abnormalities.
Obviously, current categorization systems for CC are complexity and diversity, due to different principles of classification (Figure 1). From the perspective of ophthalmologists, these systems possess both strengths and weaknesses and their respective areas of application are different.
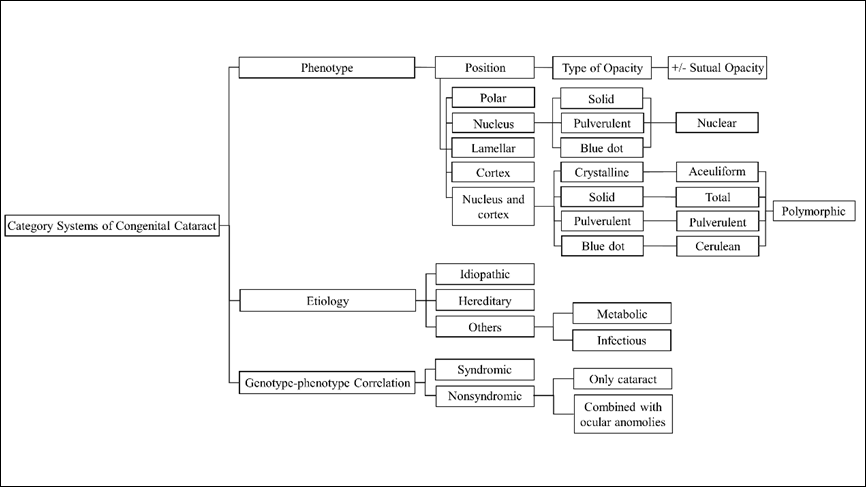
Figure 1 Diffirent categorization systems of congenital cataract
The morphology and locations of CC exhibit considerable variation.[15] In terms of morphology, CC can be classified as solid, pulverulent, or blue dot, among others. With respect to anatomical locations, CC can be categorized as nuclear, cortical and more, as depicted in Figure 2. Multiple classifications have been the morphology or locations of CC.[4,15-19] Reddy et al.[20] presented a relatively comprehensive classification method by integrating the morphology and locations of opacities within the lens. According to this classification, CC is divided into anterior and posterior polar, nucleus, lamellar, nucleus and cortex, and cortex based on their respective locations. Among these, the nucleus and cortex can be further subcategorized into solid, pulverulent, blue dot and crystalline based on the type of opacity. This classification system is widely employed in clinical practice due to its convenience in facilitating direct differential diagnosis by ophthalmologists.
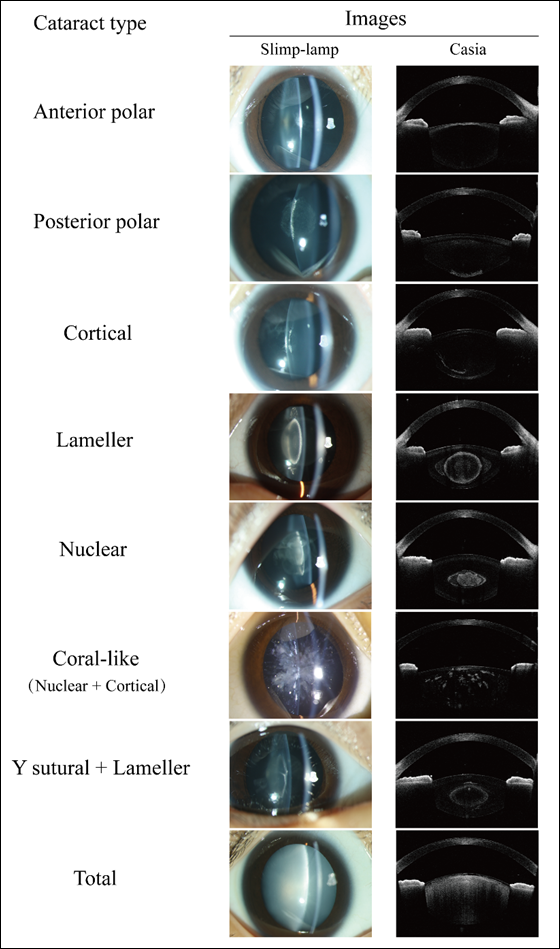
Figure 2 Slit-lamp and Swept-source anterior segment Optical Coherence Tomography (AS-OCT, Casia) images of different congenital cataract morphologies
Many reports have described the distribution of different morphological types of CC. In Marshall et al.’s study,[21] nuclear cataract was the most prevalent type (30.5%) among their cohort, followed by lamellar cataract (25.9%) and posterior lenticonus (19.0%). In Haargaard et al.’s research,[6] nuclear/zonular cataract was identified as the most frequent morphological type, accounting for 34% of all cases. Similarly, Yang et al.[22] observed a significantly elevated prevalence of nuclear cataract (48.4%), followed by total cataract (22.1%) and posterior polar cataract (13.8%). In general, nuclear cataracts are the predominant morphological type within the CC cohort. Moreover, the distribution of CC morphology differs between bilateral and unilateral cases. Nagamoto et al.[23] observed that total cataract (29.3%), nuclear cataract (19.4%), and lamellar cataract (18.5%) were prevalent in bilateral cases, whereas unilateral cases predominantly exhibited posterior subcapsular/polar cataract (34.7%), nuclear cataract (23.5%), and total cataract (21.8%). Long et al.[19] arrived at a comparable conclusion, with nuclear cataract (35.8%), total cataract (32.3%), and lamellar cataract (11.1%) being predominant among bilateral cases, while unilateral cases mostly presented with polar cataract (42.3%), total cataract (32.3%), and nuclear cataract (17.7%). This phenomenon is thought to potentially associated with gene mutations or embryonic development.
The morphology of cataracts plays an important role in predicting surgical outcomes. Marshall et al.[21] observed significant variations in that postoperative visual acuity among different cataract types, with lamellar and posterior lenticonus groups exhibiting the most favorable outcomes, while nuclear group demonstrated relatively poorer results. Li et al.[24] documented suboptimal visual outcomes in patients with bilateral total congenital cataracts, with 93/176 (52.8%) eyes achieving a postoperative best-corrected visual acuity (BCVA) of < 20/200. Kim et al.[25] found that postoperative BCVA for posterior polar cataracts (0.34±0.12 logMAR) surpassed that of nuclear (0.39±0.22 logMAR) and total (0.58±0.42 logMAR) cataracts. Mistr et al.[26] reported surgery outcomes of 30 eyes with posterior polar cataracts and 32 eyes with posterior lenticonus, revealing that 84% and 68% of eyes in the respective groups achieved acuities better than 20/40. Chen et al.[27] reported similar findings, with 21/30 (70%) of eyes diagnosed with posterior polar cataracts or lenticonus achieving a BCVA better than 20/40. Conversely, Kekunnaya et al.[28] found poorer visual outcomes in cases of posterior lenticonus, with 29/59 (49%) of eyes exhibiting a BCVA worse than 20/100 or eccentric fixation 6 months postoperatively. Generally, total and nuclear cataracts are related to unsatisfactory visual prognosis, requiring aggressive surgical intervention as early as possible. The varying prognoses observed in posterior polar cataracts and lenticonus indicate that the influence of other factors such as age at surgery, potential fundus lesions and preoperative visual acuity.[27-28] Treatment strategies should be tailored according to the specific morphology of cataract.
While this classification is a simple and useful tool in clinical practice, it is not without its drawbacks. Firstly, due to the absence of a detailed classification standard, ophthalmologists may encounter difficulty in precisely discerning the specific locations or morphology of CC sometimes. Furthermore, this classification seems to overlook other coexisting ocular conditions. Common ocular comorbidities in congenital cataracts include aniridia, microphthalmos, persistent fetal vasculature (PFV), nystagmus, and strabismus. Nagamoto et al.[23] found that compared to unilateral cases, bilateral cases exhibited a notably high frequency of nystagmus, while in unilateral cases, strabismus emerged as the most prevalent associated ocular diseases. Additionally, strabismus, nystagmus and microphthalmos were frequently observed in cases of total cataract, while PFV often presenting in eyes affected by posterior subcapsular/polar cataracts. Haargaard et al.[6] highlighted the frequent occurrence of PFV in unilateral cases, while noting that microphthalmos and microcornea were more prevalent in bilateral cases. Ocular comorbidities hold the potential to influence the formulation of treatment strategies and patients’ prognosis. For example, in cases of PFV, the fetal vessels may strongly adhere to the posterior capsule, resulting in preoperative or intraoperative posterior capsule rupture[29] and intraoperative hemorrhage[4]. To avoid severe complications, comprehensive preoperative examinations are required, and surgical procedures must be meticulously planned. Furthermore, individuals with CC may also present various multisystem disorders, such as Down’s syndrome[30], Oculo-Facio-Cardio-Dental (OFCD) syndrome[31], Trisomy 21, Lowe syndrome and many others[32], all of which have the potential to significantly impact patients’ prognosis.
Recently, there have been rapid advancements in anterior segment measurement instruments, such as anterior segment Optical Coherence Tomography or Pentacam connected to a digital Scheimpflug rotational camera. Based on the locations of lens opacities shown in slit-lamp and Pentacam examinations, Liu et al.[33] established a classification for CC into four groups: total, anterior, interior, and posterior cataracts. In this classification, total cataract is defined as opacity of entire lens, while interior cataract refers to denotes opacity of the interior lens without involvement of anterior or posterior capsules. Anterior and posterior cataracts indicate lens opacities with involvement of anterior or posterior capsules, respectively. This classification method, being straightforward, is both simple and practical for ophthalmologists to employ in in clinical settings. Using this classification, they explored the relationship between locations of CC and other anterior segmental characteristics. Although significant corneal astigmatism (CA) was found in all types of CC, anterior cataract showed the strongest association with CA. As the locations of CC became more posterior, the value of CA decreased. Additionally, anterior chamber depths (ACD) varied across the different types, with interior and posterior cataract had greater ACD, while total and anterior cataracts exhibited shallower ACD. Other studies also focused on the abnormal anterior segment structure in CC. Han et al.[34] observed the distribution of anterior corneal astigmatism (ACA), posterior corneal astigmatism (PCA), and total corneal astigmatism (TCA) of patients with CC. They also found large CA in patients with CC, with 70.3% of cataractous eyes (378/538) displaying an ACA exceeding1.25 D, and 72.8% of cataractous eyes (392/538) exhibiting a TCA exceeding1.25 D. Among cataractous eyes, 70.6% of cataractous eyes (380/538) had PCA ranging from 0.25 to 0.75 D. In addition to anterior segmental characteristics, Liu and coworkers[35] further into the link between visual function and ocular structure in patients diagnosed with posterior cataract. The discovered that compared with their contralateral healthy eyes, cataractous eyes exhibited a prolonged peak time of P100 of PVEP-60’, along with a diminished amplitude of P100 of PVEP-60’.
In contrast to the traditional 2-dimensional image-based classification, this modified classification system leverages 3-dimensional images for categorization of CC, yielding heightened precision in clinical applications. Furthermore, it facilitates the anticipation of potential anterior segment irregularities based on the locations of lens opacities and offers more pragmatic guidance in formulating treatment approaches for ophthalmologists. Nonettheless, despite its simplicity and practicality in clinical settings, this method overlooks the varied morphologies and severity gradients of CC, alongside potential ocular or systemic comorbidities. Moreover, in instances where the morphology of CC is intricate, this classification method may not be applicable.
The etiology of cataracts presents a range of causes. Cataracts can be categorized into several types based on their origins, including idiopathic, hereditary, metabolic, infectious cataracts.[36] Reports indicate that idiopathic cataract represent the most prevalent form, accounting for up to 50% of all the cases.[6,37-38] Patients diagnosed as idiopathic CC can have other ocular or systemic disorders, thereby requiring the exclusion of hereditary, infectious and other etiologies. Notably, unilateral cases are more likely to be idiopathic in comparison to bilateral instances. While the risk factors of idiopathic CC remain unconfirmed, earlier reports have suggested a potential correlation with prenatal or perinatal causative factors.[37] Following idiopathic cataracts, hereditary cataract represent the second common type, comprising over 20% of all the cases.[39] Hereditary modes of CC include autosomal dominant, autosomal recessive, and X-linked recessive inheritance, with autosomal dominant inheritance being the most prevalent.[40] Hereditary cataracts demonstrate considerable clinical and genetic heterogeneity.[41-42] Additionally, metabolic disorders such as galactosemia, hypoglycemia, hypocalcemia can lead to the development of cataracts.
Galactosemia arises from a deficiency in any of the three enzymes, including galactokinase, galactose-1-phosphate uridy1 transferase, or uridine diphosphate-galactose-4-epimerase.[43] Galactose is converted into galactitol in the lens, leading to the accumulation of galactitol and subsequent osmotic changes. Initially, the lens exhibit soil-droplet opacification, which may progress to cortical or nuclear opacification. This process is reversible if galactose is promptly excluded from the diet during the early stages. Additionally, intrauterine infections, particularly those caused by toxoplasma gondii, rubella virus, syphilis, cytomegalovirus, herpes simplex virus (TORCH), can lead to CC. Among these, rubella virus infection is the most prevalent. Besides cataract, the clinical manifestations of congenital rubella syndrome include glaucoma, pigmentary retinopathy, heart disease, and brain dysfunction. However, the precise pathogenesis of CC resulting from intrauterine rubella infection remains elusive. Nguyen et al.[44] reported that the viral infection within the ciliary body may underlie the development of cataract.
To a certain extent, classifying cataracts based on their etiology can serve as a guide for clinicians in determining treatment strategies. For instance, patients diagnosed with hereditary CC, may require genetic testing and counseling for their families. Individuals with galactosemia would benefit from a galactose-free diet. In cases where cataracts are caused by rubella virus, clinicians should consider the patients’ general health to rule out severe systemic comorbidities. However, achieving the precise etiological diagnosis of CC can be challenging at times. In clinical practice, etiology classification is not used independently but rather combined with other CC classifications to facilitate improved diagnosis and treatment.
With the emergence and application of genetic technology, researchers have embarked on the exploration of potential genetic abnormalities associated with hereditary cataract. Presently, the literature reports an association of more than 100 genes with both nonsyndromic or syndromic hereditary cataract.[40,45] Hereditary cataracts show significant variation within and between families. It is noteworthy that a single mutation can give rise to diverse forms of cataracts, while identical types of cataracts can result from mutations in different genes. To elucidate the suspected genetic causes based on phenotype, some scholars have categorized the implicated genes into four groups, guided by the genotype-phenotype correlation: the genes implicated in syndromic cataracts, genes associated specifically with congenital cataracts within syndromic cases, genes exclusively associated with cataracts, and genes associated with both cataract and eye anomalies.[11]
The extent to which currently known genes can account for the occurrence of cataracts has varied across different studies. More than 50 genes have been identified in association with nonsyndromic cataract, including crystalline genes, membrane transport protein genes, developmental regulator and transcription factor genes, cytoskeletal protein genes, and transmembrane protein genes.[46] Crystalline genes are particularly prominent in nonsyndromic hereditary cataracts, demonstrating a range of phenotypic variations. Berry et al.[47] reported 10 different crystallin variants in CRYAA, CRYBA1, CRYBB1, CRYGD, and CRYGC, with nuclear or lamellar phenotypes being predominant. A variant in CRYBB2 was found to be associated with nuclear-sutural phenotype.[48] Rogaev et al. identified variant in the γ-crystallin gene.[49] that may contribute to non-nuclear phenotypes. Cai et al.[50] discovered a variant in the CRYGD gene associated with coralliform phenotype. Disease-causing variants in CRYAB, CRYBA2, CRYBA3, CRYBA4, and CRYGS have been reported, and these variants exhibit association with various cataract phenotypes.[48,51] Mutations in membrane transport protein genes can result in a diverse array of phenotypes, including total, nuclear, Y-sutural with nuclear, pulverulent, lamellar, lamellar sutural, pearl box, coralliform, punctate, Coppock-like cataracts.[11,52-53] A Variant in LIM2 gene, in particular, has been linked to membranous cataract.[54] Mutation in developmental regulator and transcription factor genes, such as of PAX6 and HSF4, are associated with a wide range of phenotypes, including lamellar, punctate, coralliform, anterior and posterior polar, nuclear cataracts.[52,55] Whereas, variants in PITX3 are associated with narrower range of cataract phenotypes, including posterior polar, total, and nuclear phenotypes.[55] It is worth noting that variants in some of the above-mentioned genes can give rise to other ocular anomalies. For instance, Zin et al.[56] reported that a missense variant in CRYBB3 could lead to both cataract and microphthalmia. Gao et al.[57] found that variant in CRYGD could cause cataract along with nystagmus. When cataracts co-occur with abnormalities in the other organs, it results in syndromic cataracts. More than 200 syndromes have been associated with hereditary cataracts, exhibiting diverse morphologies and genetic causes.[11,46,58]
Undeniably, this classification system has the potential to enhance our understanding of the genotype-phenotype correlation in hereditary cataracts, thereby aiding in the genetic counseling for patients and their families. However, due to its clinical and genetic heterogeneity of the condition, there are challenges in establishing a precise genotype-phenotype correlation of hereditary cataract. Therefore, it is difficult to solely rely on complex phenotypes for genetic classification of hereditary cataracts.
There exist multiple categorization systems for CC, some of which are utilized in genetic and basic researches. However, the practical application of CC categorization systems in clinical practice is limited, such as appearing as only baseline data in clinical studies or neglecting eye and systemic symptoms other than cataract itself in clinical practice. At present, obtaining precise data on morphology, etiology, and genetics within a specific classification remains challenging, and not all classifications reliably predict the visual outcomes of patients. Therefore, to comprehensively characterized CC conditions and facilitate in treatment decisions and prognostic assessment, it is important for clinicians to adopt a comprehensive approach by utilizing multiple CC classifications instead of relying solely on a specific one. By incorporating various classification systems, clinicians can gain a more complete understanding of the condition, leading to improve management strategies and patient care.
In addition, future research should focus on integrating dimensions for classifying CC. The phenotypic characteristics of CC patients, including ocular biometry of anterior and posterior segments, usually often exhibit variations among individuals. The current CC categorizations systems primarily rely on single standards, which may not adequately capture the comprehensive ocular characteristics of patients. Therefore, it is important to develop new categorization systems that incorporate multiple dimensions. For example utilizing ocular biometry and considering the conditions of anterior and posterior segments as classification criteria. Furthermore, it is crucial to explore the correlation between this new classification system and the visual prognosis of children with CC. By doing so, we can enhance the clinical value and applicability of the classification system. The ultimate objective of future research is to improve the clinical and scientific utility of CC categorization systems, leading to more accurate diagnoses, effective treatment approaches, and better outcomes for patients.
Correction notice
NoneAcknowledgement
Our heartfelt appreciation to Ms. Yin Qiuxia for her dedicated efforts in meticulously improving the language and grammar of this article with her precious time.Author Contributions
(I) Conception and design: Zhenzhen Liu(II)Administrative support: Zhenzhen Liu
(III) Provision of study materials or patients: Yingshi Zou and Yunqian Li
(IV) Collection and assembly of data: Yingshi Zou and Yunqian Li
(V) Data analysis and interpretation: Yingshi Zou and Yunqian Li
(VI) Manuscript writing:All authors
(VII) Final approval of manuscript:All authors
Funding
This work was supported by the Joint Funding Project of Municipal Schools (Colleges) of Science and Technology Program of Guangzhou, China (2023A03J0188) and the Guangzhou Municipal Science and Technology Project (202201011815).The funding organizations had no role in the following aspects: design and conduct of the study; the collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and the decision to submit the manuscript for publication.