


HIGHLIGHTS
1. Critical Discoveries and Outcomes
• In chronic genetic models of retinal degeneration, such as rd10 mice, colony-stimulating factor 1 receptor(CSF1R) targeted interventions have been shown to modulate microglial activation and slow progressive photoreceptor loss. These models mainly reflect long-term neuroinflammatory remodeling driven by inherited defects.
2. Methodological Innovations
• Using an acute N-methyl-N-nitrosourea (MNU) induced degeneration model, this study demonstrates thatCSF1R-dependent microglial depletion is associated with preservation of photoreceptor structure and scotopic visual function. These effects are accompanied by broad transcriptional changes involving inflammatory signaling, mitochondrial and metabolic pathways, and neuron-related programs. The findings extend current understanding by identifying microglia as key regulators of early injury-driven degenerative dynamics.
3. Prospective Applications and Future Directions
• These results define CSF1R-dependent myeloid signaling as a tractable mechanistic pathway in acute retinal degeneration and provide a conceptual framework for future studies investigating context-specific modulation of neuroinflammatory responses.
Introduction
Retinal degeneration (RD), including retinitis pigmentosa, represents a leading cause of irreversible blindness worldwide and is defined by progressive photoreceptor loss.[1-2] Despite advances in gene therapy, stem cell transplantation, and anti-apoptotic interventions, current therapeutic options remain limited in efficacy and are often accompanied by safety concerns.[3-5] Increasing evidence highlights neuroinflammation and defective clearance of dying neurons as central drivers of disease progression, positioning them as critical nodes for therapeutic intervention.[6-8] In this context, retinal microglia, the resident immune cells, emerge as key modulators of both neuroinflammatory responses and phagocytic activity, making them an attractive target for strategies aimed at preserving neuronal integrity.[9-10]
Despite increasing recognition of microglial involvement in retinal degeneration, their functional impact remains highly context-dependent. In several experimental models, microglial depletion has been associated with reduced photoreceptor loss and attenuated disease progression,[11-13] whereas in other settings the absence of microglia exacerbates neuronal damage, suggesting divergent roles in injury responses.[14-15] These inconsistencies highlight an incomplete understanding of how microglia influence degenerative trajectories under different pathological conditions. In particular, their specific contribution to acute toxic retinal injury remains poorly defined. The N-methyl-N-nitrosourea (MNU) model,[16-17] characterized by rapid primary photoreceptor degeneration followed by secondary inflammatory remodeling, provides a valuable system to interrogate microglial function during early stages of injury-driven retinal degeneration.
Pharmacological inhibition of colony-stimulating factor 1 receptor (CSF1R) has become a widely adopted strategy for transient and efficient depletion of retinal microglia, given the essential role of CSF1R signaling in microglial survival. Previous studies using CSF1R inhibitors such as PLX5622 have demonstrated that modulation of myeloid activation can influence disease progression in chronic, genetically driven models of retinal degeneration, including the rd10 mouse.[18] However, whether CSF1R-dependent microglial responses similarly shape degenerative outcomes in acute photoreceptor injury paradigms remains unclear.
To address this gap, we investigated the impact of CSF1R inhibition-mediated microglial depletion in the MNU model, a reproducible acute toxic model that enables interrogation of early degeneration dynamics. By integrating structural imaging, functional assessment, and transcriptomic profiling, this study aims to define the net contribution of microglia to inflammatory and degenerative processes following acute photoreceptor loss. The findings extend current understanding of microglial roles beyond chronic remodeling contexts and provide additional insight into neuroimmune regulation in injury-driven retinal degeneration.
Materials and methods
Animals and treatments
All animal experiments were approved by the Animal Care and Use Committee of Zhongshan Ophthalmic Center and conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Male C57 BL/6J mice aged 6-8 weeks (GemPharmatech Co., Ltd) were used. To deplete microglia, mice were fed PLX5622 formulated AIN-76A diet (1,200 ppm; SYSE Bio-tec Co., Ltd, Changzhou, China) for 2 weeks prior to and throughout MNU modeling period. The MNU-induced retinal degeneration model was established by a single intraperitoneal injection of MNU (40 mg/kg), freshly prepared at 5 mg/ml (0.5% w/v) in PBS. All mice were euthanized by cervical dislocation 5 days post-injection. Eyes were immediately enucleated and processed for downstream analyses. Unless otherwise specified, one eye per mouse was used for each experimental endpoint to ensure independence of biological replicates.
Scotopic electroretinogram (ERG) recording and optical coherence tomography (OCT) analysis
Scotopic ERG was performed as previously described.[19] The amplitudes of a- and b- waves were quantified across a series of flash intensities using the Celeris-Diagnosys system. 5 mice were analyzed per experimental group. OCT images were acquired using SPECTRALIS-OCT (Heidelberg, Germany) equipped with a mouse objective lens. Scanning protocols and ONL thickness measurements were performed following the previously described procedure.[19] B-scan images centered on the optic nerve head were obtained under standardized acquisition settings. ONL thickness was quantified at 10 predefined equidistant locations (5 positions on each side of the optic nerve head at 100 µm intervals), and values were averaged to obtain a single measurement per eye. 4 mice were included in each group.
Retinal fundus imaging
Following anesthesia and pupil dilation, retinal fundus images were acquired using a Micron IV fundus camera (Phoenix Research Labs, Pleasanton, CA, USA) with the mouse objective centered on the optic disc. 4 mice were analyzed per experimental group.
Immunofluorescence staining
For immunofluorescence analysis of microglial depletion, enucleated eyes were fixed in 4% paraformaldehyde for 1 h at room temperature, followed by retinal dissection and preparation as whole-mount flat mounts to allow comprehensive spatial visualization. Retinas were incubated with anti-Iba-1 (Wako, 1:400) and anti-TMEM119 (Oasis Biofarm, 1:400). Microglial density (cells/mm2) was quantified using predefined regions of interest (ROIs) distributed across central and peripheral retinal areas in all 4 quadrants. Measurements from individual ROIs were averaged to generate a single representative value per retinal region for each animal. 3 mice were analyzed per group.
Hematoxylin & Eosin (HE) staining and histological analysis
HE staining was conducted as previously described.[20] To quantify retinal degeneration, the number of photoreceptor nuclei in the ONL was measured using ImageJ software. 4 mice were analyzed per experimental group.
RNA extraction, library construction, sequencing, and bioinformatic analysis
Total RNA from PLX5622+MNU and CD (control diet) +MNU retinas was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. RNA-seq library construction and sequencing were performed by Novogene (Beijing, China) on an Illumina NovaSeq platform to generate 150-bp paired-end reads. Low-quality reads were filtered to obtain clean reads, which were then aligned to the mouse reference genome (GRCm38, NCBI) using HISAT2 v2.0.5 with the corresponding gene model annotation; gene-level counts were obtained with featureCounts v1.5.0-p3. Differentially expressed genes (DEGs) were identified using the DESeq2 R package, with | log2(fold change(FC)) | ≧ 1 and P value < 0.05 as thresholds. Gene ontology (GO)/Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were conducted using the clusterProfiler R package. Gene-set enrichment analysis (GSEA) was performed via the GSEA software (http://www.gsea-msigdb.org/gsea/index.jsp).
Western blot analysis
Retinal tissues were homogenized in RIPA lysis buffer supplemented with protease inhibitors. Equal amounts of total protein were separated by SDS-PAGE and transferred onto PVDF membranes. After blocking, membranes were incubated overnight at 4 °C with primary antibodies against IL-1β (Cell Signaling Technology, 12242S), C1qa (Abcam, ab71940), C3 (Santa Cruz Biotechnology, sc-58926), and β-actin (Cell Signaling Technology, 3700S). Membranes were then incubated with HRP-conjugated secondary antibodies and developed using standard chemiluminescence methods. Three retinas (one retina per mouse) were analyzed for each experimental group.
Statistical analysis
Data were presented as mean ± standard error of the mean (SEM). Normality of data distribution was assessed prior to parametric testing. Comparisons between two groups were conducted using unpaired two-tailed Student’s t-test. For comparisons involving more than two groups, one-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparisons test was applied. ERG responses across graded flash intensities were analyzed using two-way repeated-measures ANOVA (factors: experimental group and stimulus intensity), followed by Tukey’s post hoc test for multiple comparisons. The experimental unit was defined as one eye from each animal unless otherwise specified. The P values < 0.05 were considered statistically significant.
Results
CSF1R inhibition protects retinal structure in MNU-induced degeneration
To determine the contribution of microglia to retinal degeneration, C57BL/6J mice were fed CSF1R inhibitor PLX5622 beginning 14 days before MNU adminstration and continuing throughout the 5-day MNU treatment period to achieve microglial depletion (Figure 1A). Retinal flat-mount immunofluorescence with TMEM119 and Iba-1 co-staining demonstrated near-complete depletion of TMEM119+/Iba-1+ retinal microglia after 19 days of PLX5622 treatment, which was further supported by quantitative analysis of microglial density in central and peripheral retinal regions (Figure 1B). We also confirmed that the PLX5622 diet alone did not alter retinal structure by OCT analysis. Strikingly, comprehensive structural analyses revealed robust protection in PLX5622+MNU retinas compared with CD+MNU retinas. Fundus photography demonstrated a marked reduction in the characteristic white lesions, which are hallmarks of outer retinal (photoreceptor layer) injury and are prominently observed in CD+MNU retinas (Figure 1C). Optical coherence tomography (OCT) demonstrated a significant preservation of outer nuclear layer (ONL) thickness (Figure 1C). Histological analysis (H&E staining) corroborated the protective effect, showing maintained retinal architecture and an increased number of ONL nuclei in the PLX5622+MNU group (Figure 1C). Collectively, these structural analyses demonstrate that PLX5622 treatment, accompanied by marked retinal microglial depletion, significantly preserved retinal structure in the MNU-induced degeneration model.
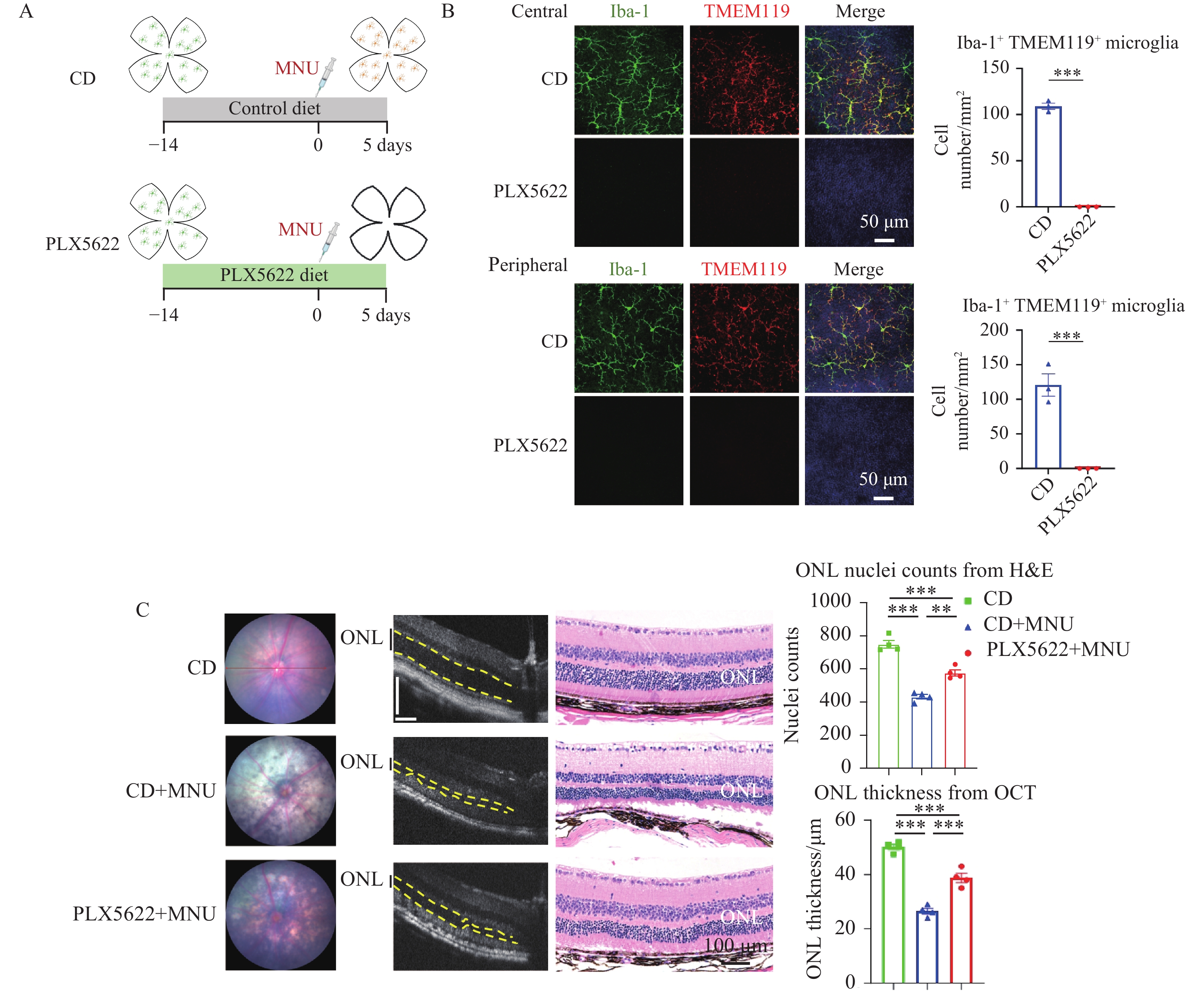
(A) Schematic of experimental timeline showing continuous PLX5622 administration starting 14 days prior to and maintained throughout the 5-day MNU treatment period. (B) Representative retinal flat-mount confocal images stained for TMEM119 and Iba-1 show near-complete depletion of TMEM119+/Iba-1+ retinal microglia after 19-day PLX5622 treatment. Quantification of microglial density (cells/mm2) in central and peripheral retinal regions is shown on the right. Scale bar = 50 μm. n = 3 retinas per group (one retina per mouse). Unpaired two-tailed Student’s t-test. (C) Representative fundus images (left) showed reduced characteristic white lesions in PLX5622+MNU retinas compared to CD+MNU retinas. OCT images (middle) illustrated PLX5622 treatment preserved ONL thickness in MNU-induced retinal degeneration mice; scale bar=110 μm. HE staining images (right) confirmed the preserved ONL architecture and the increased photoreceptor nuclei density. Scale bar = 100 μm; n = 4 eyes per group (one eye per mouse); one-way ANOVA followed by Tukey’s post hoc test. Data are shown as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. CD: control diet; ONL: outer nuclear layer.
CSF1R inhibition protects retinal function in MNU-induced degeneration
To assess the functional impact of microglial depletion, full-field scotopic electroretinography (ERG) was performed. Overall, ERG responses analysis revealed significant preservation of retinal function in PLX5622+MNU-treated mice compared with CD+MNU-treated mice. Compared with CD group, the PLX5622-only group showed no significant change in retinal function, whereas both the a-wave amplitudes (reflecting photoreceptor activity) and b-wave amplitudes (reflecting inner retinal activity, primarily bipolar and Müller cells) were significantly attenuated in the CD+MNU group. In contrast, these amplitudes were markedly preserved across multiple stimulus intensities in the PLX5622+MNU group (Figure 2A, B).
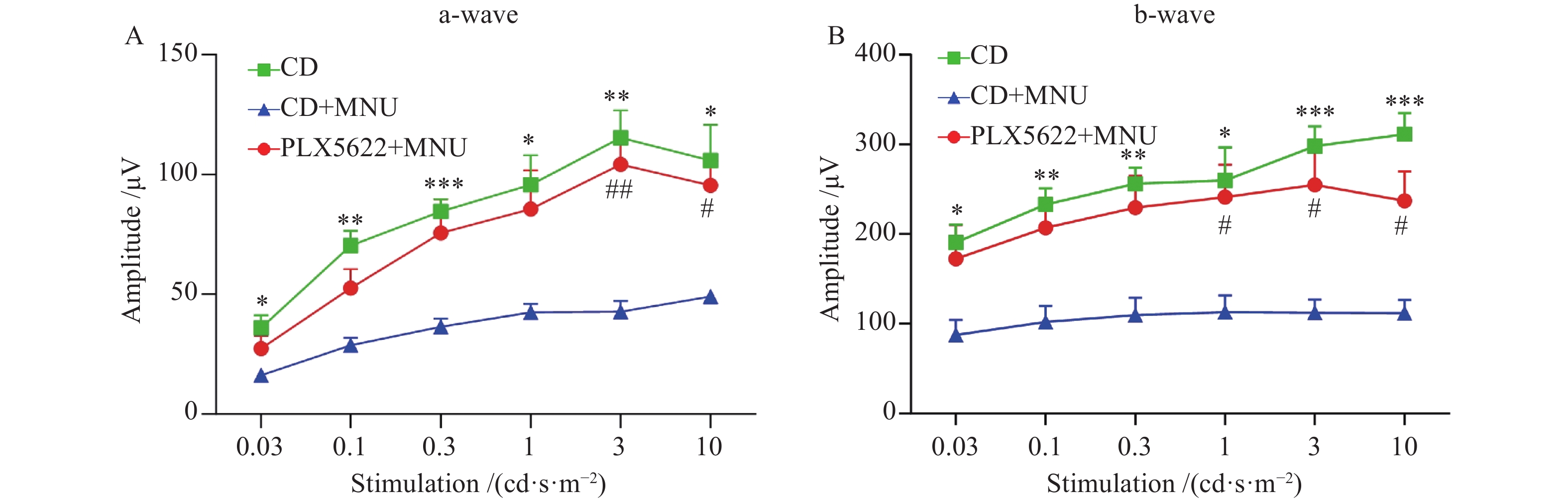
(A-B) Scotopic ERG amplitudes (a-wave and b-wave) were markedly reduced after MNU treatment compared with untreated controls (CD), whereas PLX5622 treatment significantly preserved ERG responses in MNU-challenged mice. n = 5 eyes (one eye per mouse), two-way repeated-measures ANOVA (group × stimulus intensity) followed by Tukey’s multiple-comparison test. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, CD vs CD+MNU; #P < 0.05, ##P < 0.01, PLX5622+MNU vs CD+MNU.
CSF1R inhibition is associated with reduced inflammatory signatures and altered metabolic pathways.
To further investigate the influence of CSF1R inhibition on MNU-injured retinas, we performed transcriptome sequencing of PLX5622+MNU retinas and CD+MNU retinas. Principal component analysis (PCA) demonstrated clear separation between two groups (Figure 3A), indicating substantial transcriptomic remodeling. The volcano plot showed significant downregulation of microglial cone genes (e.g., Cx3cr1, Tmem119, P2ry12, Trem2, C1q family) (Figure 3B). Further functional enrichment (GO/KEGG) analysis of upregulated DEGs revealed pathways related to antioxidant stress responses, including melanin biosynthetic process, melanin metabolic process, phenol-containing compound biosynthetic process, and glutathione metabolism,which may also be linked to retinal pigment epithelium (RPE) functional regulation (Figure 3C, E). GO and KEGG enrichment of downregulated DEGs demonstrated enriched pathways encompassing innate immune activation (including myeloid leukocyte activation and neutrophil extracellular trap formation), cytokine signaling (including regulation of interleukin-6 production), and inflammation (including chemokine signaling, complement and coagulation cascade) (Figure 3D, E). GSEA enrichment analysis further corroborated upregulation of mitochondrial energy metabolism-related pathways, including respiratory electron transport and oxidative phosphorylation (Figure 3G, H). Western blot analysis confirmed reduced expression of selected inflammatory mediators, including IL-1β, C1qa, and C3, following PLX5622 treatment (Figure 3I). Collectively, these findings indicate that CSF1R inhibition is associated with reduced inflammatory signatures, together with altered antioxidant-related and metabolic pathways.
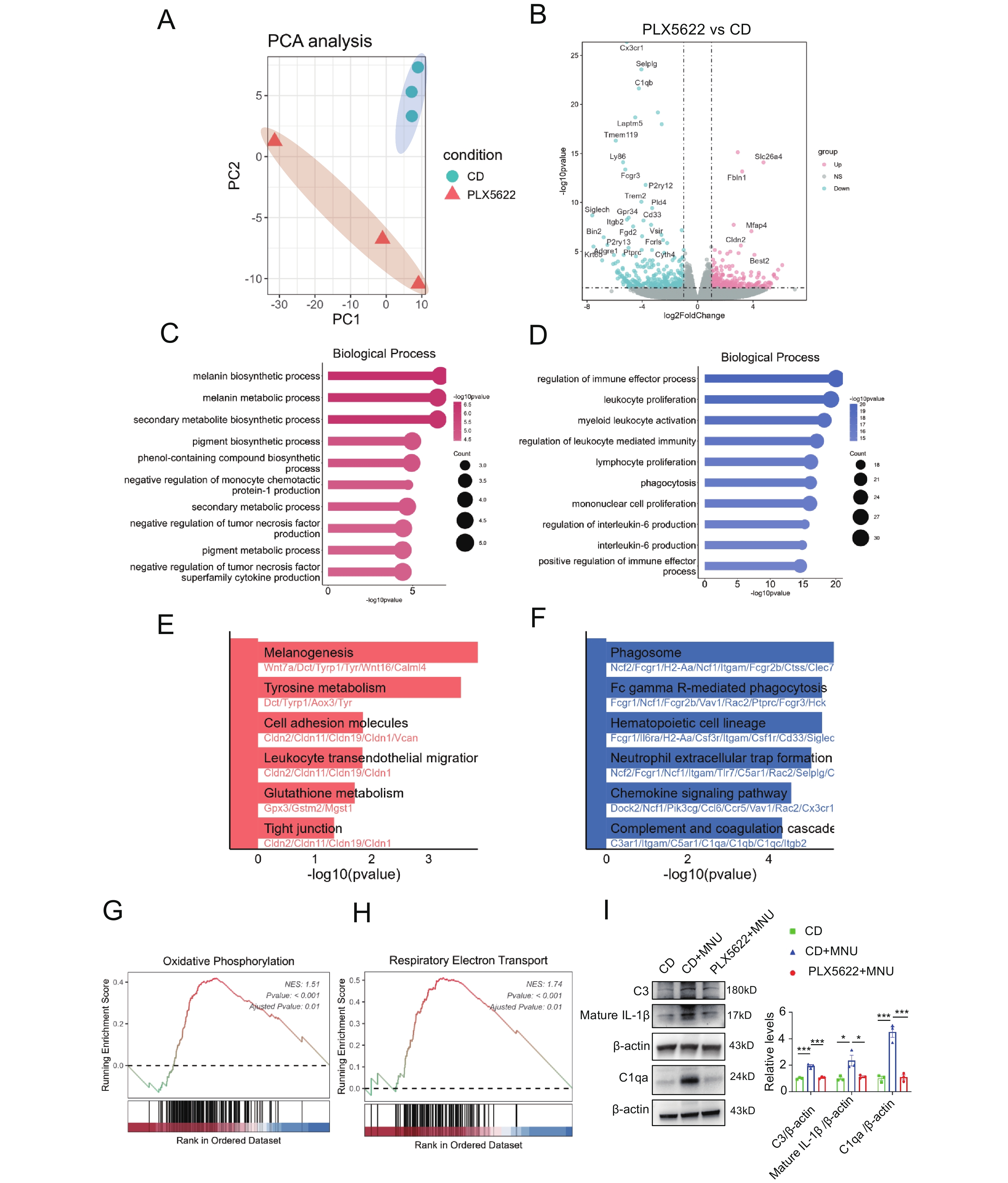
(A) PCA plot showed distinct transcriptome profiles between two groups. n = 3 replicates for each group. (B) Volcano plot [x-axis = log2FoldChange, y-axis=-log10pvalue] showing 569 differentially expressed genes (DEGs). Red dots indicate the upregulated genes, and the blue dots indicate the downregulated genes. (C–F) GO and KEGG enrichment analyses of DEGs selected using the thresholds of log2FC ≥ 1 and p-value < 0.05. Top enriched terms/pathways are ranked by -log10(p-value). (C) GO biological process enrichment of upregulated DEGs. (D) GO biological process enrichment of downregulated DEGs. (E) KEGG pathway enrichment of upregulated DEGs. (F) KEGG pathway enrichment of downregulated DEGs. (G-H) GSEA analysis showed positive enrichment of respiratory electron transport and oxidative phosphorylation pathways. (I) Representative Western blot images and quantification of C3, C1qa, and mature IL-1β in the CD, CD+MNU, and PLX5622+MNU groups. n = 3 retinas per group (one retina per mouse). One-way ANOVA followed by Tukey’s post hoc test. *P < 0.05; **P < 0.01; ***P < 0.001.
CSF1R inhibition is associated with modulation of neuron-related and macroglia gene programs
To characterize transcriptomic changes linked to neuronal and macroglial responses, we further analyzed GO and GSEA-enriched pathways. GO analysis of upregulated DEGs in PLX5622+MNU retinas revealed enrichment of neurodevelopmental and synaptic signaling-related processes, including pathways associated with positive regulation of nervous system development, regulation of neuroblast proliferation, neuromuscular synaptic transmission, and cholinergic synaptic transmission (Figure 4A). In contrast, downregulated DEGs were enriched in pathways related to the neuronal apoptotic process (Figure 4B). In parallel, GO analysis of downregulated DEGs showed enrichment of macroglia-related pathways, including astrocyte differentiation and astrocyte activation, whereas GSEA further demonstrated downregulation of glial cell activation-related programs (Figure 4B, C). Together, these findings indicate that CSF1R inhibition is associated with coordinated transcriptomic shifts involving neuronal survival-related programs and attenuation of macroglial activation signatures.
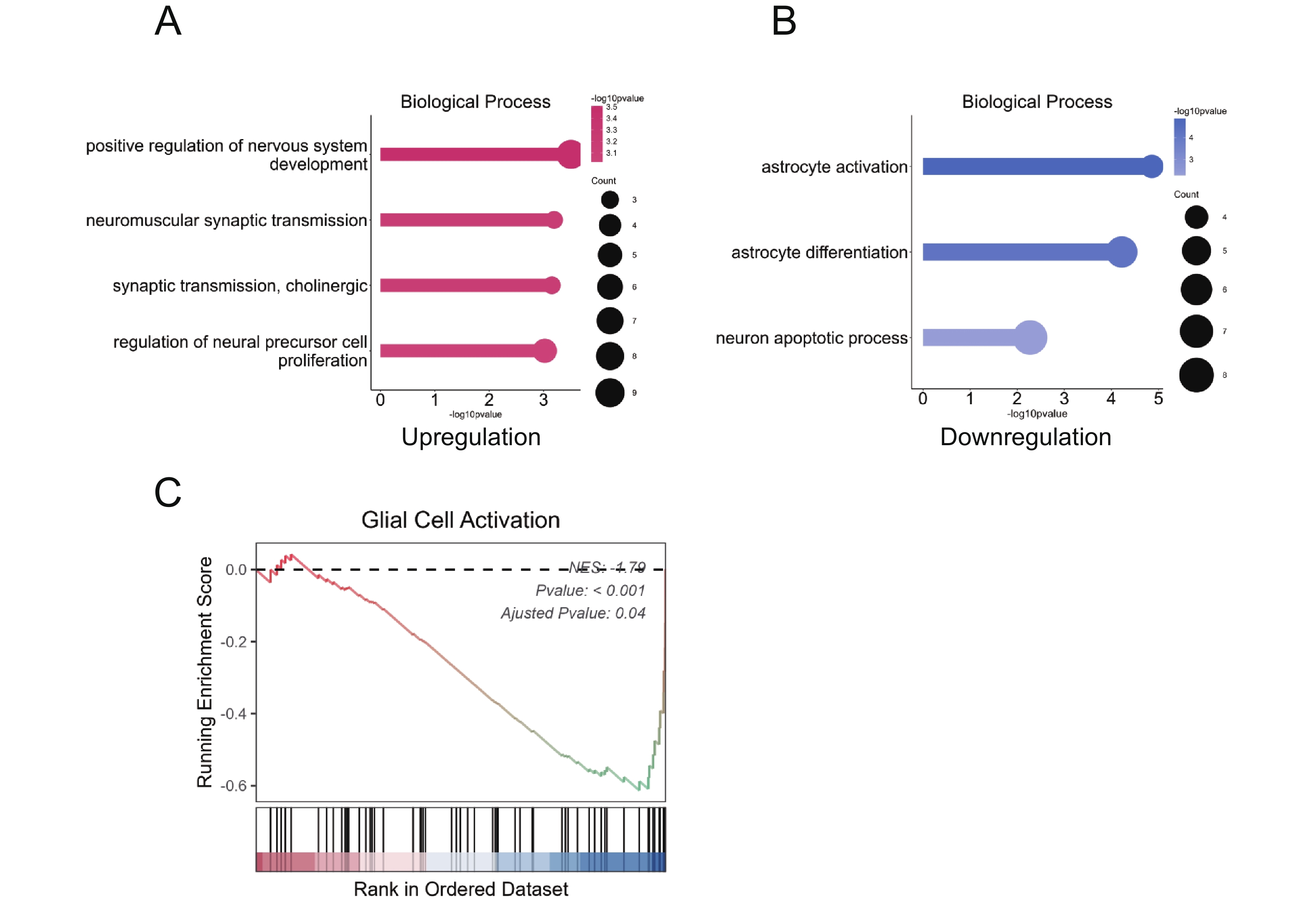
(A) GO biological process enrichment for upregulated DEGs, highlighting pathways associated with neural development and synaptic function. (B) GO biological process enrichment for downregulated DEGs, showing suppression of pathways linked to astrocyte activation, astrocyte differentiation and neuron apoptosis. (C) GSEA plot significant negative enrichment of the ‘glial cell activation’ gene set.
Discussion
In this study, pharmacological CSF1R inhibition with PLX5622 resulted in effective retinal microglial depletion and was associated with attenuation of MNU-induced degeneration at structural, functional, and transcriptomic levels. PLX5622-treated eyes exhibited reduced fundus lesions, preservation of outer nuclear layer thickness and photoreceptor nuclear density, and maintenance of scotopic ERG responses across stimulus intensities. These observations support a contributory role of microglia-associated inflammatory processes in shaping retinal injury during acute toxic degeneration.
Our findings are broadly consistent with recent study by Wu et al., who reported that CSF1R inhibition with PLX5622 mitigates neurotoxic myeloid activation and preserves photoreceptors in the rd10 model of inherited retinitis pigmentosa, a chronic degeneration paradigm driven by Pde6b mutation.[18] However, the MNU model represents a mechanistically distinct paradigm characterized by rapid photoreceptor apoptosis and temporally compressed inflammatory responses. By examining CSF1R-dependent myeloid modulation in this acute injury context, the present study extends current understanding of microglial involvement beyond chronic remodeling processes and provides convergent evidence across complementary models of retinal degeneration.
As a DNA-alkylating agent, MNU induces a rapid and relatively synchronized wave of photoreceptor apoptosis, followed by the release of injury-associated signals that activate resident microglia and amplify secondary inflammation.[21] This feature makes the MNU model particularly useful for dissecting the early neuroinflammatory responses that follow acute neuronal injury. Although it does not fully recapitulate the chronic course of inherited retinal degeneration, the model captures an important component shared by several clinically relevant conditions involving abrupt photoreceptor loss, such as acute retinal detachment, severe photic injury, and autoimmune retinopathy.[11,22-24] In these settings, the initial neuronal injury may already be established, whereas subsequent microglia-driven inflammatory remodeling may further exacerbate tissue damage. Our findings therefore suggest that early modulation of microglial responses after acute photoreceptor injury may help interrupt this secondary injury cascade and limit retinal degeneration.
Our transcriptomic analyses provided additional mechanistic insight. RNA-seq of PLX5622+MNU retinas compared to CD+MNU retinas revealed profound remodeling of the global retinal microenvironment. This included reduced inflammatory and innate immune gene signatures, together with enrichment of antioxidant-related terms and mitochondrial/metabolic-related pathways. Additional enrichment analyses also indicated neuron-associated and synaptic-related gene programs among upregulated DEGs, whereas glial activation-related signatures were reduced. Western blot analysis further showed that a set of inflammatory mediators (IL-1β, C1qa, and C3) were elevated in MNU-injured retinas and attenuated after PLX5622 treatment. Together, these findings suggest that PLX5622 treatment suppresses inflammatory responses associated with retinal degeneration and is accompanied by broader transcriptomic changes in metabolic- and neuron-associated programs.
Several limitations should be acknowledged. CSF1R inhibition influences multiple myeloid populations, and thus the protective effects observed in this study cannot be attributed exclusively to resident retinal microglial depletion. In our experiments, whole-mount immunostaining for TMEM119 and Iba-1 demonstrated near-complete loss of resident retinal microglia following prolonged PLX5622 treatment, supporting effective target engagement. However, infiltrating peripheral monocytes/macrophages were not systematically quantified after MNU injury. Therefore, the observed protective phenotype is best interpreted as reflecting CSF1R-dependent modulation of myeloid responses, and future lineage-resolved analyses will be required to clarify the relative contributions of resident and recruited myeloid cells. Second, the prolonged PLX5622 pretreatment was intended to achieve efficient microglial depletion and should be interpreted primarily as a mechanistic paradigm rather than a therapeutic regimen. Whether similar effects can be achieved within clinically relevant intervention windows remains to be determined. Finally, although bulk transcriptomic analysis identified coordinated neuron- and macroglia-related changes, cell type-specific mechanisms require validation using higher-resolution approaches.
In summary, our findings indicate that CSF1R-dependent modulation of microglial responses can influence inflammatory and degenerative processes during acute photoreceptor injury. In the MNU model, effective microglial depletion was associated with reduced neuroinflammation and preservation of retinal structure and visual function. These results support a contributory role of microglia in shaping early degeneration dynamics in toxic retinal injury. Further studies are warranted to determine context-appropriate intervention strategies, including the timing, duration, and extent of CSF1R inhibition, as well as the potential consequences of prolonged microglial depletion.
Correction notice
None
Acknowledgements
We thank the staff of the Core Facilities at the State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center for their technical support.
Author Contributions
(I) Conception and design: Chang He
(II) Administrative support: Chang He, Xialin Liu
(III) Provision of study materials or patients: Chang He
(IV) Collection and assembly of data: Hong Zhou, Biyan Ni, Tian Zhou, Ziqi Yang, Yang Zhou, Jingpeng Li, Minglu Ma, Huaicheng Wang, Peng An, Qingbo Guo, Xiaojing Lin, Shiya Lin, Huiyi Xu
(V) Data analysis and interpretation: Hong Zhou, Biyan Ni, Chang He
(VI) Manuscript writing: All authors
(VII) Final approval of manuscript: All authors
Conflict of Interests
None of the authors has any conflicts of interest to disclose. All authors have declared in the completed the ICMJE uniform disclosure form.
Patient consent for publication
None
Ethics approval and consent to participate
All animal experiments were approved by the Animal Ethics Committee of Zhongshan Ophthalmic Center (No.2020-102), and were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Data availability statement
None
Open access
This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License(CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited(including links to both the formal publication through the relevant DOI and the license).