基于高通量测序分析鉴别视网膜病变中的神经纤维瘤病
'%20fill='white'%20fill-opacity='0.01'/%3e%3cmask%20id='mask0_3477_29692'%20style='mask-type:luminance'%20maskUnits='userSpaceOnUse'%20x='0'%20y='0'%20width='16'%20height='16'%3e%3crect%20id='&%23232;&%23146;&%23153;&%23231;&%23137;&%23136;_2'%20x='16'%20width='16'%20height='16'%20transform='rotate(90%2016%200)'%20fill='white'/%3e%3c/mask%3e%3cg%20mask='url(%23mask0_3477_29692)'%3e%3cpath%20id='&%23232;&%23183;&%23175;&%23229;&%23190;&%23132;'%20d='M14%205L8%2011L2%205'%20stroke='%23333333'%20stroke-width='1.5'%20stroke-linecap='round'%20stroke-linejoin='round'/%3e%3c/g%3e%3c/g%3e%3c/svg%3e)
关键词
摘要
全文
文章亮点
1. 关键发现
• 通过高通量测序分析,鉴别临床初诊为其他视网膜病变的神经纤维瘤病,并发现合并有多种视网膜病变的罕见病例。2. 已知与发现
• 神经纤维瘤病累及视网膜的改变复杂多样,临床初诊为不同视网膜疾病。• 高通量测序分析可识别以不典型视网膜病变首诊的神经纤维瘤病,早期预警隐匿的严重神经系统改变。
• 罕见病例中神经纤维瘤病可同时合并多种不同的视网膜病变。
3. 意义与改变
• 基于高通量测序的基因变异分析,有助于识别以不典型视网膜病变首诊的神经纤维瘤病,从而早期预警和干预隐匿的严重神经系统改变。神经纤维瘤病(neurofibromatosis, NF)是一种由于基因缺陷引起的多系统受累的常染色体显性遗传病,包括3种类型,即1型神经纤维瘤病(neurofibromatosis type 1, NF1)、NF2相关神经鞘瘤病(既往称为2型神经纤维瘤病,neurofibromatosis type 2, NF2)和其他神经鞘瘤病[1-3],其中,NF1和NF2相关神经鞘瘤病是最常见的类型[4]。
1 资料和方法
1.1 一般资料
1.2 方法
2 结果
2.1 本研究中NF1和NF2基因变异的致病性分析
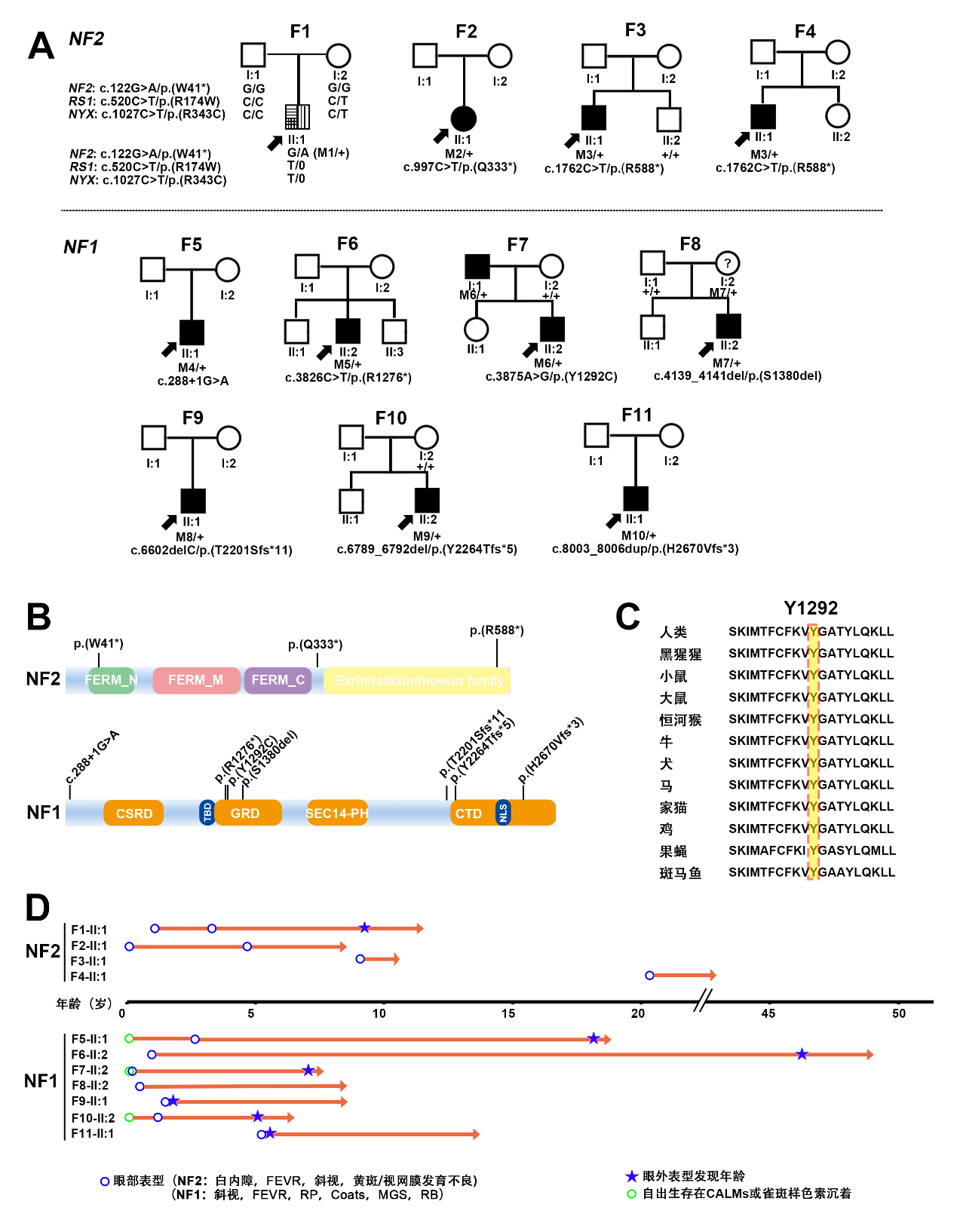
图1 本研究中11个携带NF1和NF2致病变异的家系
|
表1 本研究在11个家系中发现10个NF1和NF2变异 Table 1. Ten variants in NF1 and NF2 identified in 11 families with different eye conditions in our cohort |
||||||||
|
变异编号 |
外显子 |
碱基改变 |
氨基酸改变 |
家系数 |
ACMG/AMP分类 |
ACMG/AMP证据 |
HGMD |
首次报道 |
|
NF2 (NM_000268.3) |
|
|
|
|
|
|
||
|
M1 |
2 |
c.122G>A |
p.(W41*) |
1 |
P |
PVS1, PS2, PM2 |
DM |
Evans DG, Trueman L, Wallace A, et al. Genotype/phenotype correlations in type 2 neurofibromatosis (NF2): evidence for more severe disease associated with truncating mutations[J]. J Med Genet, 1998, 35(6): 450-455. DOI: 10.1136/jmg.35.6.450 |
|
M2 |
10 |
c.997C>T |
p.(Q333*) |
1 |
P |
PVS1, PM2, PM6 |
DM |
Sestini R, Vivarelli R, Balestri P, et al. Neurofibromatosis type 2 attributable to gonosomal mosaicism in a clinically normal mother, and identification of seven novel mutations in the NF2 gene[J]. Hum Genet, 2000, 107(4): 366-371. DOI: 10.1007/s004390000378. |
|
M3 |
16 |
c.1762C>T |
p.(R588*) |
2 |
P |
PVS1_Moderate, PM2, PM6 |
DM |
Van Hout CV, Tachmazidou I, Backman JD, et al. Exome sequencing and characterization of 49, 960 individuals in the UK Biobank[J]. Nature, 2020, 586: 749-756. DOI: 10.1038/s41586-020-2853-0. |
|
NF1 (NM_000267.3) |
|
|
|
|
|
|
||
|
M4 |
3 |
c.288+1G>A |
/ |
1 |
P |
PVS1, PM2, PM6 |
DM |
A M John, M Ruggieri, R Ferner, M Upadhyaya.A search for evidence of somatic mutations in the NF1 gene[J].J Med Genet. 2000, 37(1):44-9. DOI: 10.1136/jmg.37.1.44. |
|
M5 |
28 |
c.3826C>T |
p.(R1276*) |
1 |
LP |
PVS1, PM6 |
DM |
R A Heim, L N Kam-Morgan, C G Binnie, et al.Distribution of 13 truncating mutations in the neurofibromatosis 1 gene[J]. Hum Mol Genet. 1995, 4(6):975-81. DOI: 10.1093/hmg/4.6.975 |
|
M6 |
29 |
c.3875A>G |
p.(Y1292C) |
1 |
LP |
PS1, PP1, PP3 |
DM |
Ruen Yao, Tingting Yu, Yufei Xu, et al. Clinical Presentation and Novel Pathogenic Variants among 68 Chinese Neurofibromatosis 1 Children[J]. Genes (Basel). 2019, 10(11):847. DOI: 10.3390/genes10110847 |
|
M7 |
31 |
c.4139_4141del |
p.(S1380del) |
1 |
LP |
PVS1, PM2, PM4 |
/ |
本研究 |
|
M8 |
43 |
c.6602delC |
p.(T2201Sfs*11) |
1 |
P |
PVS1, PM2, PM6 |
/ |
本研究 |
|
M9 |
45 |
c.6789_6792del |
p.(Y2264Tfs*5) |
1 |
P |
PVS1, PM2, PM6 |
/ |
本研究 |
|
M10 |
54 |
c.8003_8006dup |
p.(H2670Vfs*3) |
1 |
P |
PVS1, PM2, PM6 |
/ |
本研究 |
|
注:DM:有害变异;P:致病;LP:可能致病。 Note: DM, deleterious mutation. |
||||||||
2.2 携带NF2致病变异患者表现复杂视网膜病变
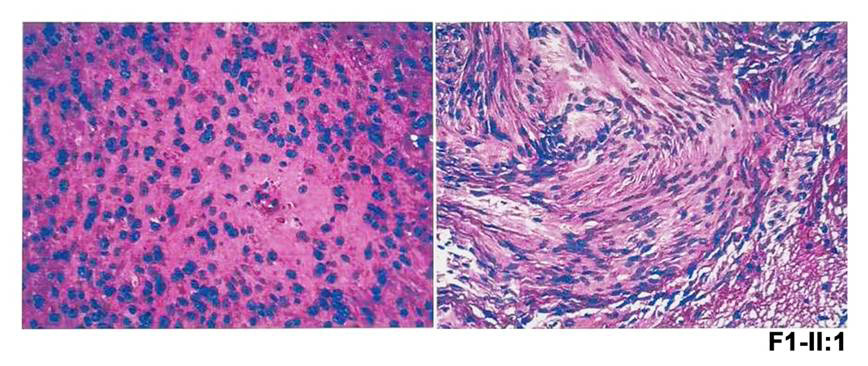
本研究中共有4个家系携带NF2基因致病变异,其临床表型各异,首诊表现均为复杂的眼部表型。其中,F1-Ⅱ:1为1例10岁男孩,该患儿3岁时因“出生后3个月起发现斜视”来诊,其右眼BCVA为0.6,左眼BCVA为指数眼前,屈光度数右眼为+0.50 DS/–1.00 DC ×80 °,左眼为–0.25 DS/–3.00 DC×10 °,眼前节未见明显异常(附图2A)。眼底检查显示右眼黄斑中央凹反光不明显;左眼视盘发育不全,黄斑区纤维增殖膜及血管从视盘区拖拽至颞侧中周部视网膜,与FEVR样改变相类似。OCT显示双眼黄斑区视网膜增厚,左眼黄斑前膜。FFA未观察到明显血管渗漏[25]。该患儿自症状起曾多次于各地眼科门诊就诊,初步诊断均疑诊为FEVR。为进一步明确其致病原因,对患儿外周血DNA行TES[17]检测,结果发现在RS1基因中存在一个半合子致病变异c.520C>T/p.(R174W),但在FEVR相关基因中并未发现致病变异,因此考虑修改该患者诊断为X连锁视网膜劈裂症(X-linked retinoschisis, XLRS)[25]。然而,该基因变异尚未能解释患儿复杂眼部改变,因此,为了进一步探讨并发视网膜病变的可能原因,对其进行了WGS检测。结果,患者除了存在RS1致病变异外,还检测到另外2个基因的致病变异,包括NYX半合子变异c.1027C>T/p.(R343C)、NF2新发变异c.122G>A/p.(W41*)(表1)。根据上述结果,在患儿10岁时对其进行了回访,包括全身皮肤观察、CFP、SLO、FAF、OCT、ERG、颅脑和脊椎MRI等系统检查。结果发现,患儿全身皮肤并未发现任何CALMs、雀斑样色素沉着等,而双眼眼底改变(图2A)与7年前相似[25]。ERG结果显示患儿视杆反应明显下降,出现b波负波改变,且视锥振幅轻度降低(附图2B),提示患儿为完全型先天性静止性夜盲(complete congenital stationary night blindness, CSNB1)。完善颅脑和脊椎MRI检查,发现患儿双侧VS,延髓内存在一扩张性髓内强化信号(考虑星形细胞瘤可能性大),C1—2、T8—L5脊柱多发神经鞘瘤(图2B),眶周组织未见明显异常(附图2A),表明患儿表型与NF2基因变异相符。因此,基于基因检测结果以及眼科和神经学的系统检查,该患儿最终诊断为NF2相关神经鞘瘤病/XLRS/CSNB1。伴随眼部变化的NF患者常存在侵袭性的中枢神经系统肿瘤[26],且NF2无义变异与NF2相关神经鞘瘤病的严重程度相关[3, 27],本研究患儿与之相符。由于延髓中的肿瘤可能存在压迫呼吸中枢的风险而危及生命,因此,该患儿转诊至神经外科,行显微手术切除肿瘤,并康复出院。术后组织病理学检查显示该肿瘤为脊髓室管膜瘤(中枢神经细胞瘤WHO Ⅱ级),伴神经鞘瘤(附图3)。针对该患儿其他病变的处理,主要是神经科随访,以及针对NF2相关神经鞘瘤病的相关眼部改变和视网膜劈裂,每年进行眼科检查,密切监测眼部变化。
此外,本研究在另外3例不同眼病且不相关的先证者中发现NF2另外两个致病变异(表1和表2)。F2-Ⅱ:1为5岁女孩,因双眼视力差首诊,检查发现双眼白内障,右眼视网膜脱离,FFA提示周边部视网膜血管毛刷样变及荧光渗漏,OCT提示左眼黄斑区视网膜前膜牵拉视网膜增厚,该患儿初诊为双眼先天性白内障,双眼FEVR,右眼视网膜脱离(图2C)。为进一步明确病因,完善基因检测,同样地,该患儿的基因检测结果并未发现FEVR相关致病基因变异,但发现NF2基因致病变异c.997C>T/p.(Q333*)。因此,结合基因检测结果(NF2基因致病变异)及OCT(左眼黄斑前膜伴视网膜错构瘤)(图2C),考虑F2-Ⅱ:1为NF2相关神经鞘瘤病。另外携带NF2基因c.1762C>T/p.(R588*)的2例先证者(F3-Ⅱ:1和F4-Ⅱ:1)则表现为黄斑萎缩和杯盘比增大(表2)。
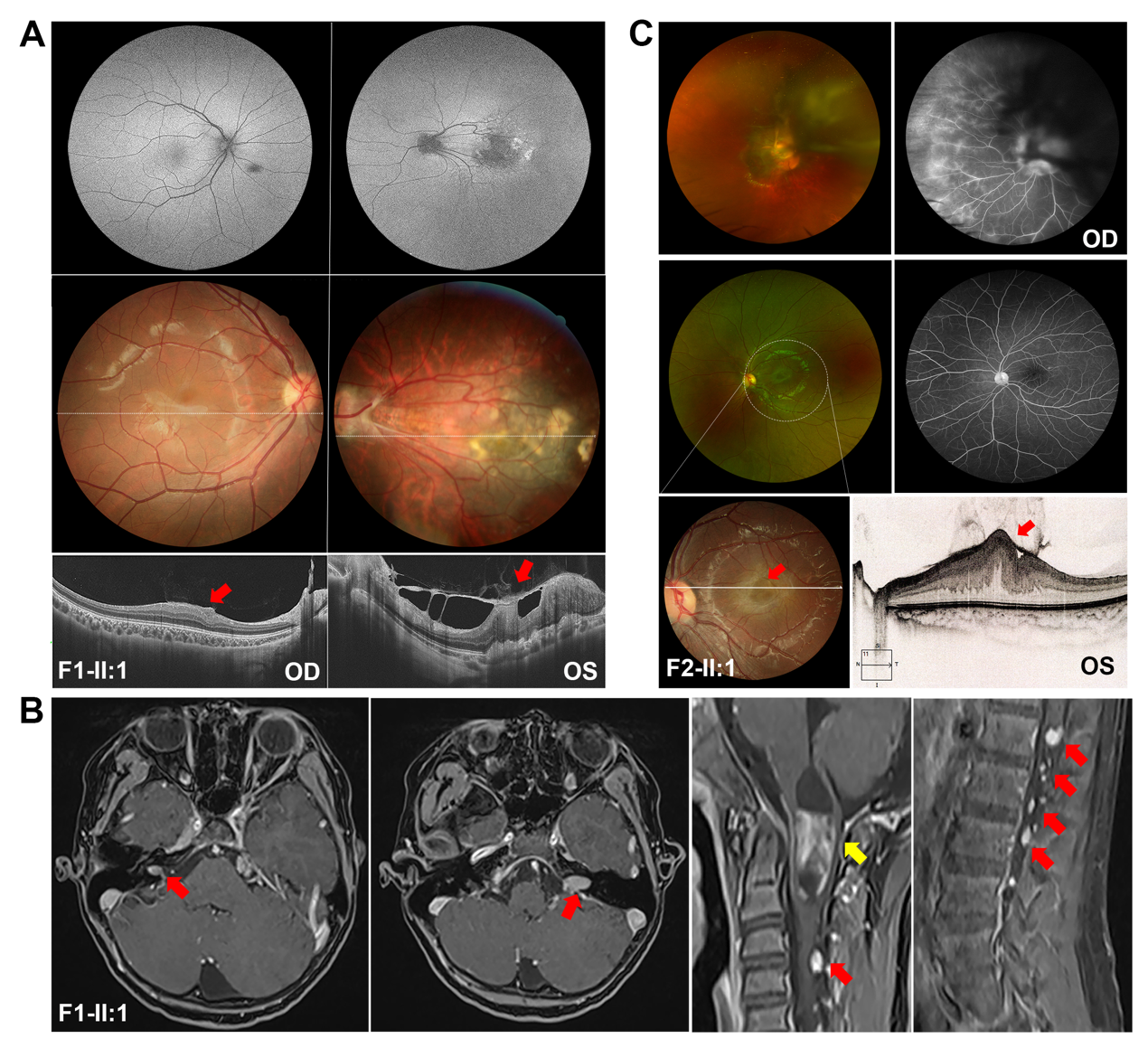
图 2 携带NF2致病变异先证者的临床表型
Figure 2 Clinical phenotypes of individuals with variants in NF2
|
表2. 本研究总11例携带NF1和NF2致病/可能致病变异的不相关先证者的临床表型 Table 2. Clinical manifestations of 11 unrelated probands with P/LP variants in NF1 and NF2
|
|||||||||
|
先证者编号 |
变异编号 |
外显子 |
碱基改变 |
氨基酸改变 |
性别 |
发病年龄 (岁)# |
眼科首诊 |
眼部表型 |
眼外表型 |
|
NF2 (NM_000268.3) |
|
|
|
|
|
|
|
||
|
F1-II:1* |
M1 |
2 |
c.122G>A |
p.(W41*) |
男 |
0.25 |
FEVR |
黄斑中央凹反光不明显(右眼);视盘发育不全,黄斑区纤维增殖膜及血管从视盘区拖拽至颞侧中周部视网膜(左眼) |
VS(双侧),脊髓室管膜瘤,多发神经鞘瘤 |
|
F2-II:1 |
M2 |
10 |
c.997C>T |
p.(Q333*) |
女 |
5.6 |
FEVR |
白内障和视网膜脱离(右眼),视网膜前膜及错构瘤(左眼) |
NA |
|
F3-II:1 |
M3 |
16 |
c.1762C>T |
p.(R588*) |
男 |
9 |
RP |
C/D=0.6 |
NA |
|
F4-II:1 |
M3 |
16 |
c.1762C>T |
p.(R588*) |
男 |
33 |
RP |
黄斑发育不良 |
NA |
|
NF1 (NM_000267.3) |
|
|
|
|
|
|
|
||
|
F5-II:1 |
M4 |
3 |
c.288+1G>A |
/ |
男 |
4.5 |
FEVR |
周边视网膜微血管异常 |
CALMs |
|
F6-II:2 |
M5 |
28 |
c.3826C>T |
p.(R1276*) |
男 |
儿童期 |
RP |
视网膜色素变性 |
雀斑样色素沉着 |
|
F7-II:2 |
M6 |
29 |
c.3875A>G |
p.(Y1292C) |
男 |
4 |
Coats |
视网膜下渗出 |
雀斑样色素沉着 |
|
F8-II:2 |
M7 |
31 |
c.4139_4141del |
p.(S1380del) |
男 |
0.6 |
MGS |
牵牛花样视盘改变 |
无 |
|
F9-II:1 |
M8 |
43 |
c.6602delC |
p.(T2201Sfs*11) |
男 |
1.3 |
OG |
突眼 |
OG,头部皮下肿块 |
|
F10-II:2 |
M9 |
45 |
c.6789_6792del |
p.(Y2264Tfs*5) |
男 |
1.4 |
斜视 |
斜视 |
CALMs,小脑神经纤维瘤浸润 |
|
F11-II:1 |
M10 |
54 |
c.8003_8006dup |
p.(H2670Vfs*3) |
男 |
6.4 |
OG |
突眼 |
OG |
|
备注:FEVR:家族性渗出性玻璃体视网膜病变;VS:前庭神经鞘瘤;RP:视网膜色素变性;MGS:牵牛花综合征;CALMs:牛奶咖啡斑;OG:神经胶质瘤;NA:未获得。 *F1-II:1基于基因检测及表型分析,最终诊断为NF2相关神经鞘瘤病/XLRS/CSNB1。 |
|||||||||
|
Note: M, male; F, female; FEVR, Familial exudative vitreoretinopathy; ERM, epiretinal membrane; VS, vestibular schwannomas; CHRRPE, combined hamartoma of the retina with or without RPE; RD, retinal detachment; RP, retinitis pigmentosa; MD, macular dystrophy; CALMs, Café-au-lait macules; MGS, Morning glory disc; OG, optic glioma; NA, not available. |
|||||||||
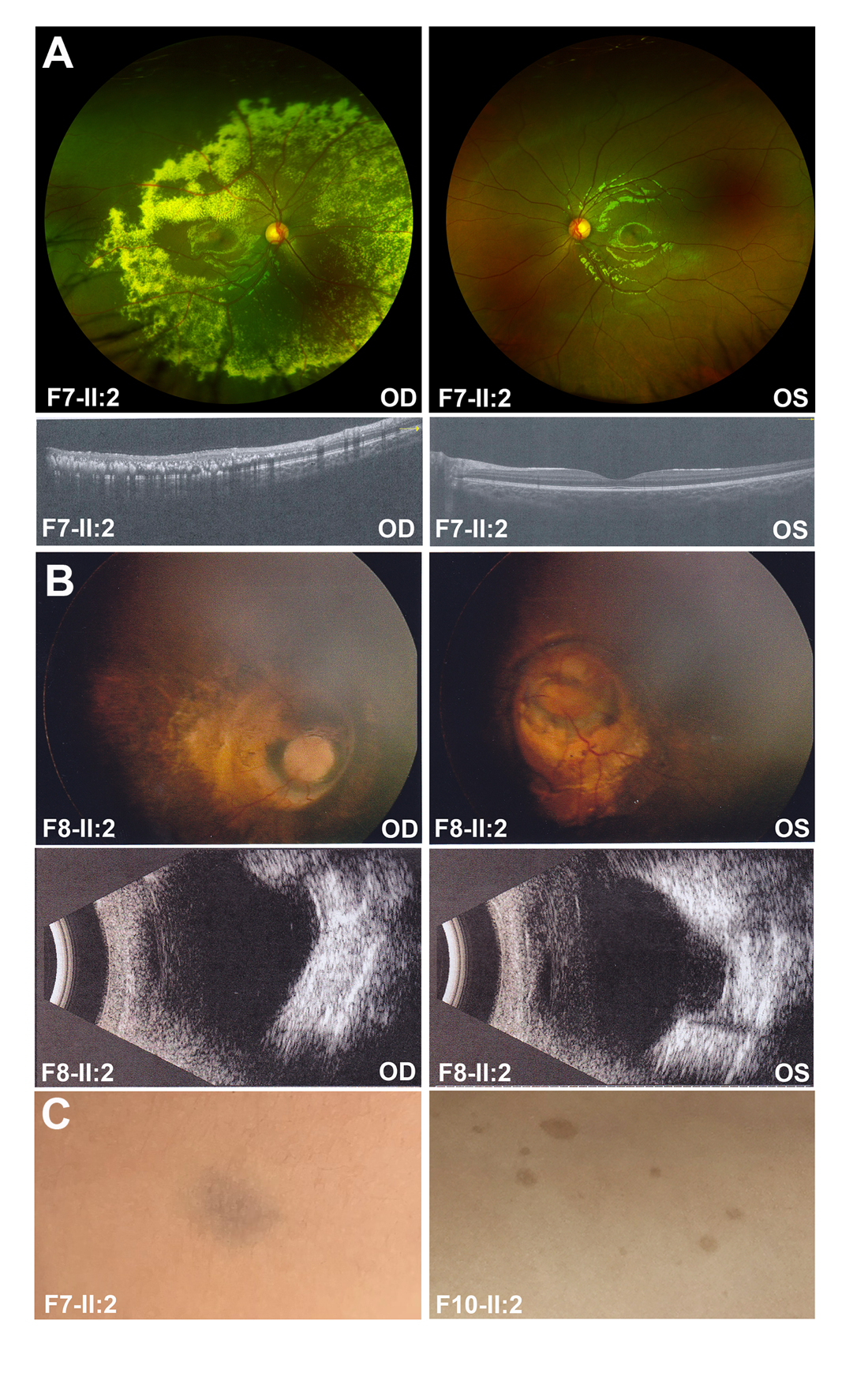
2.3 携带NF1致病/可能致病变异患者的临床表型
3 讨论
利益冲突
开放获取声明
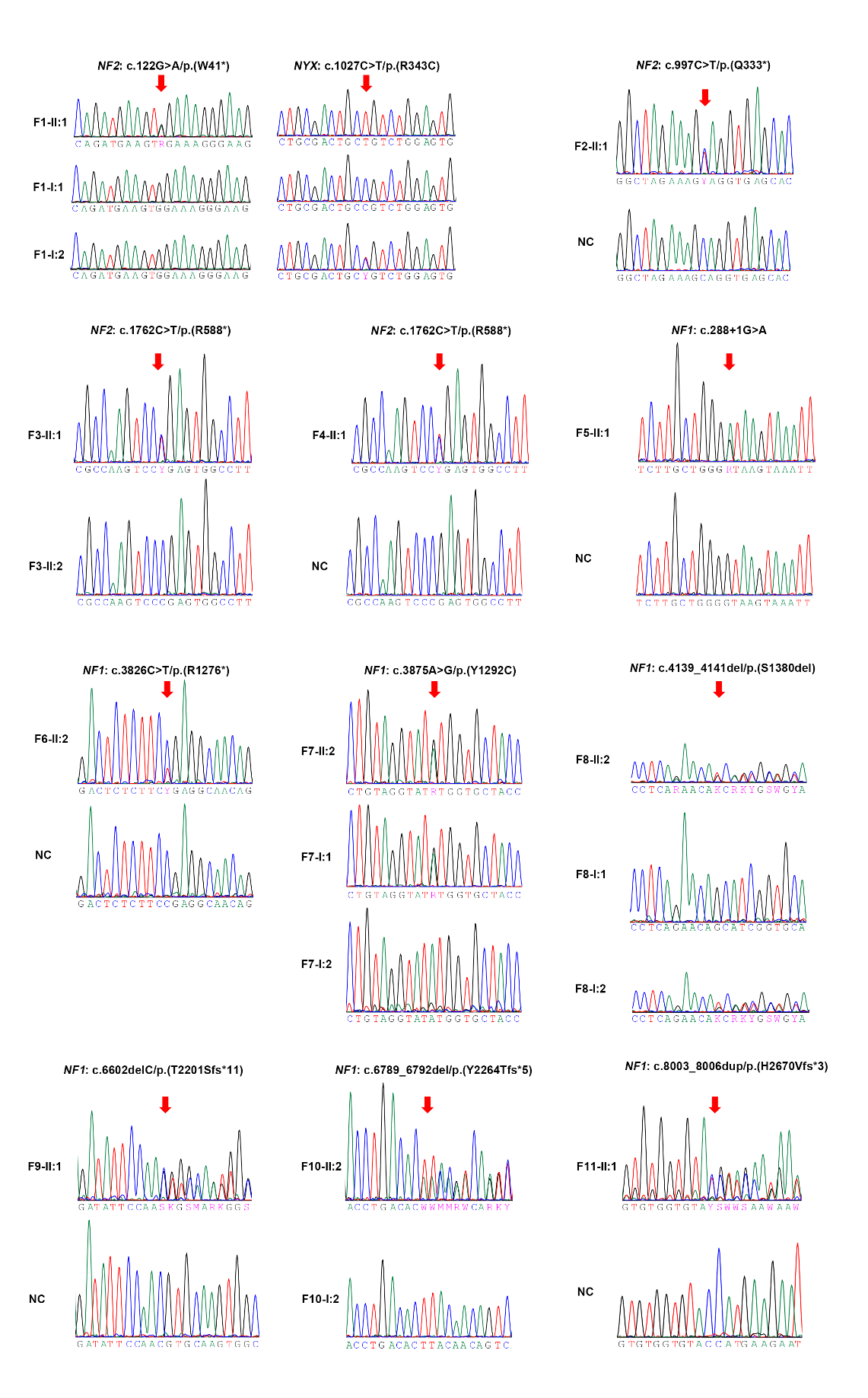
附图 1 本研究中携带NF1和NF2致病/可能致病变异的11个家系的Sanger测序图
Supplementary figure 1 Sanger sequencing chromatograph of 11 families with P/LP variants in NF1 and NF2 in this study
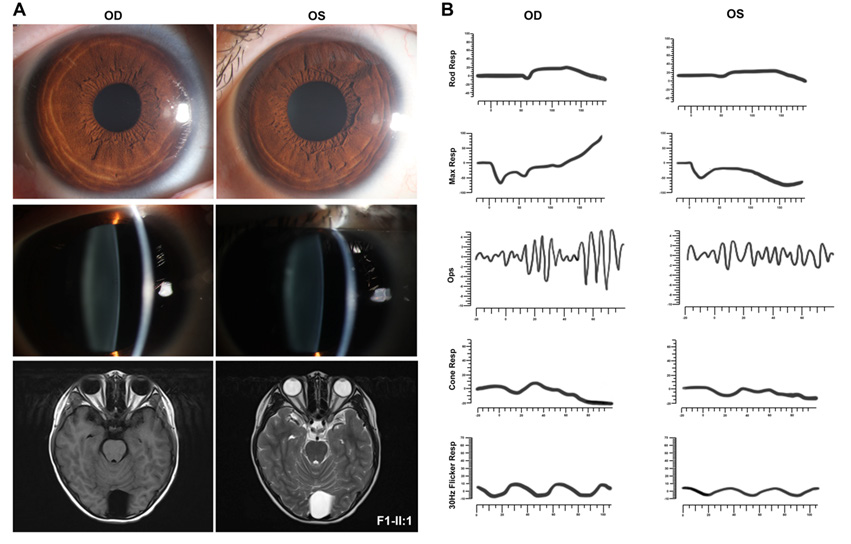
锥细胞反应轻度下降。
(A) Ocular anterior segment photograph in F1-Ⅱ:1 was with normal limit, while the orbital MRI showed no abnormality in orbital tissue.
(B) ERG showed significantly reduced rod responses and a negative waveform under bright white, with mildly reduced cone amplitude in F1-
Ⅱ:1.
基金
This work was supported by the National Natural Science Foundation of China (82171056);the Guangdong Basic and Applied Basic Research Foundation (2022A1515111060);the Science and Technology Planning Projects of Guangzhou (SL2024A03J00525);the Fundamental Research Funds of the State Key Laboratory of Ophthalmology