分子视角下的IgG4相关眼病:从自身免疫到纤维化
阅读量:1484
DOI:10.12419/24093002
发布日期:2025-09-28
作者:
胡睿 ,李光宇
展开更多 '%20fill='white'%20fill-opacity='0.01'/%3e%3cmask%20id='mask0_3477_29692'%20style='mask-type:luminance'%20maskUnits='userSpaceOnUse'%20x='0'%20y='0'%20width='16'%20height='16'%3e%3crect%20id='&%23232;&%23146;&%23153;&%23231;&%23137;&%23136;_2'%20x='16'%20width='16'%20height='16'%20transform='rotate(90%2016%200)'%20fill='white'/%3e%3c/mask%3e%3cg%20mask='url(%23mask0_3477_29692)'%3e%3cpath%20id='&%23232;&%23183;&%23175;&%23229;&%23190;&%23132;'%20d='M14%205L8%2011L2%205'%20stroke='%23333333'%20stroke-width='1.5'%20stroke-linecap='round'%20stroke-linejoin='round'/%3e%3c/g%3e%3c/g%3e%3c/svg%3e)
关键词
IgG4相关性眼病
自身免疫
纤维化
分子视角
摘要
IgG4相关性眼病(immunoglobulin G4-related ophthalmic disease, IgG4-ROD)是一种与IgG4阳性浆细胞浸润和血清IgG4水平升高相关的系统性疾病。该病的眼部表现包括泪腺、眼眶脂肪、眶下神经、眼外肌和眼睑的受累,且常伴有炎症和纤维化过程。在分子水平上,IgG4-ROD涉及多种免疫细胞和细胞因子的相互作用,包括辅助性T细胞1(T helper cell 1, Th1)、辅助性T细胞2(T helper cell 2, Th2)、调节性T细胞 (regulatory T cells, Tregs)和滤泡辅助性T细胞(follicular helper T cell, Tfh),以及由它们分泌的白细胞介素4(interleukin 4, IL-4)、白细胞介素13(interleukin 13, IL-13)和转化生长因子β(transforming growth factor-β, TGF-β)等。这些分子通过促进B细胞活化和IgG4的产生,以及纤维化过程的发生,共同参与了IgG4-ROD的发病机制。诊断依赖于组织病理学特征,治疗通常包括糖皮质激素和免疫抑制剂,旨在控制免疫介导的炎症和纤维化,减轻症状并防止器官损伤。随着对IgG4-ROD在分子层面发病机制认识的深入,治疗策略将不断优化,进而为患者提供更为精准和有效的治疗方案,从而改善患者预后,提高患者生活质量。本文基于现有研究,总结并阐述了与IgG4-ROD致病有关的关键分子及信号通路,从分子视角对IgG4-ROD的自身免疫、纤维化及二者之间的联系与转变进行综述。
全文
文章亮点
1. 关键发现
• 本综述从分子视角系统阐述了免疫球蛋白 G4 相关眼病 (immunoglobulin G4-related ophthalmic disease, IgG4-ROD) 从自身免疫反应向纤维化发展的关键机制。研究指出,IgG4-ROD 以组织中 IgG4+ 浆细胞浸润和血清 IgG4 水平升高为典型特征,但其直接致病作用尚未明确,更可能是异常免疫应答的继发表现。多种免疫细胞参与疾病发生,其中 CD4+T 细胞通过分泌白细胞介素 4(interleukin 4, IL-4)、白细胞介素 13(interleukin 13, IL-13) 及干扰素 γ(interferon-γ, IFN-γ) 等细胞因子,不仅促进 B 细胞活化及 IgG4 类别转换,还直接参与纤维化进程。B 细胞除产生抗体外,还可分泌血小板衍生生长因子 B(platelet derived growth factor B, PDGF-B)、的赖酰胺氧 化酶样蛋白 2(lysyl oxidase like 2, LOXL2) 及趋化因子 CC 基序趋化因子配体 (C-C motif chemokine ligand , CCL) 等因子直接激活成纤维细胞,导致胶原沉积和组织纤维化。疾病进程呈现从炎症向纤维化的动态转变,其中 M1/M2 巨噬细胞表型转换、炎性小体激活及细胞因子 ( 如 IL-4、IL-13) 的作用尤为关键。此外,滤泡辅助性T细胞与 B 细胞在异位生发中心中的相互作用进一步促进了 IgG4+ 浆细胞扩增和纤维化发展。这些发现为理解 IgG4-ROD 的分子机制提供了重要依据,也为开发针对特定免疫细胞及细胞因子的精准治疗策略指明了方向。2. 已知与发现
• IgG4 相关性眼病是一种与 IgG4 阳性浆细胞浸润和血清 IgG4 水平升高相关的系统性疾病。该病的眼部表现包括泪腺、眼眶脂肪、眶下神经、眼外肌和眼睑的受累,且常伴有炎症和纤维化过程。随着对 IgG4-ROD 在分子层面发病机制认识的深入,治疗策略将不断优化,进而为患者提供更为精准和有效的治疗方案。3. 意义与改变
• 本文基于现有的研究,总结并阐述了与 IgG4-ROD 致病有关的关键分子及信号通路,从分子视角对 IgG4-ROD 的自身免疫和纤维化,以及二者之间的联系与转变进行综述。从分子视角探究 IgG4-ROD 的发病机制可以为疾病的诊断和治疗提供帮助,从而改善患者预后,提高患者生活质量。IgG4相关性眼病(immunoglobulin G4-related ophthalmic disease, IgG4-ROD)是一种受自身免疫介导的眼部非特异性炎性病变,主要累及泪腺和眼外肌,少数发生巩膜炎、鼻泪管阻塞及眼周神经受压[1]。其主要的病理特征包括淋巴浆细胞浸润、闭塞性静脉炎和席纹状纤维化。而这种特征性的IgG4阳性浆细胞大量浸润不仅局限于眼部病变组织,在机体其他器官的病变中也有类似发现[1]。该线索将多种发生在不同部位的疾病关联起来,于2011年明确了IgG4相关疾病(immunoglobulin G4-related disease, IgG4-RD)的命名规则[2-3]。由于之前对于IgG4-RD的定义不明确,因此其发病率被明显低估。亚洲国家IgG4‑RD 预估发病率为每年(0.28~1.08)/100 000[4]。具有眼睑肿胀等典型临床表现的IgG4-ROD患者常被误诊为其他常见的眼眶疾病而耽误治疗。目前的病例报道主要来自日本、韩国及中国香港等东亚地区,但由于之前的认识不足,所以并不能得出IgG4-ROD在这些地区更高发的结论。对于IgG4-ROD的诊断和治疗,全球目前仍未有统一的标准。最新发布的《IgG4相关眼病诊治专家共识(2024版)》提出的诊断标准:1)影像学检查显示泪腺、三叉神经或眼外肌肿大,各种眼部肿块、增生性病变,鼻窦弥漫性炎症;2)血清 IgG4升高(≥1 350 mg/L);3)组织病理学检查显示淋巴细胞和浆细胞浸润,有时出现纤维化,常可见生发中心。IgG4+浆细胞满足以下标准:IgG4+细胞与 IgG+细胞的比率为 40% 或以上,或每个显微镜高倍(400倍)视野超过 50个 IgG4+细胞。当同时满足以上1、2和3时,诊断被归类为“确诊”;当满足 1 和 3 时 ,为“ 拟 诊 ”;当 满 足 1 和 2 时 ,为“疑诊”[4]。从分子视角探究IgG4-ROD的发病机制可以为疾病的诊断和治疗提供帮助,但目前该病关键分子及信号通路仍不明晰。本文基于现有的研究,总结并阐述了与IgG4-ROD致病有关的关键分子及信号通路,从分子视角对IgG4-ROD的自身免疫和纤维化,以及二者之间的联系与转变进行综述。
1 IgG4分子在疾病中的作用
IgG-ROD最明显的病理特点就是IgG4+浆细胞大量浸润。大部分患者伴有血清总IgG增高且血清IgG4比例明显上升。尽管在IgG4-RD中观察到自身反应性IgG4,但没有证据表明它们具有直接致病性。IgG4通常被认为是非炎症性免疫球蛋白,对Fc受体(Fc receptors, FcRs)和C1q具有低亲和力,诱导吞噬细胞活化、抗体依赖性细胞毒性或补体介导损伤的能力相对较弱。目前尚需阐明IgG4在疾病发展中的具体角色:是作为直接致病因素,还是仅为其他关键效应通路(如T辅助细胞和调节性T细胞因子极化)所导致的继发性表现[5]。同时也需明确其是源于组织损伤后的异常免疫应答,抑或对未知初始刺激因素的过度炎症反应,或可能是两种机制共同作用的结果。但毋庸置疑的是,正是因为观察到IgG4+浆细胞浸润这一疾病的显著特点,IgG4-ROD或者说IgG4-RD才得以被认知为一个新的疾病实体,进而有越来越多的学者对这一庞大的系统性疾病家族进行研究。2 IgG4-ROD与自身免疫
自身免疫性疾病的特征是针对自身正常身体成分出现异常免疫应答的病理状态,可导致炎症、细胞损伤或功能紊乱并伴有相应的临床表现[6]。自身抗体是致病性B细胞在靶向个体自身组织时产生的异常抗体,已被确定为自身免疫性疾病的标志,并且通常被认为是这些疾病的临床标志物。自身免疫所针对的分子成分(如蛋白质、糖类、核酸)统称为自身抗原。自身抗体可通过Ⅱ型超敏反应直接介导细胞破坏和细胞功能异常,与自身抗原形成免疫复合物(immune complex, IC)介导组织损伤。此外,自身反应性T细胞,主要是CD4+辅助性T细胞(T helper cell, Th)1和CD8+细胞毒性T细胞(cytotoxic T lymphocyte, CTL),也可在一定条件下通过Ⅳ型超敏反应引发自身免疫病。前者释放多种细胞因子引起以淋巴细胞、单核/巨噬细胞浸润为主的炎症反应,活化的自身反应性CTL对局部自身细胞有直接杀伤作用。IgG4-ROD患者常伴有受累组织和外周血中免疫细胞和细胞因子异常升高,以及对免疫抑制剂(如利妥昔单抗、奥妥珠单抗等)治疗的有效反应等表现[7-8]。病变组织中可见大量IgG4+浆细胞浸润。单细胞转录组分析和蛋白谱揭示,IgG4-RD中外周免疫细胞如单核细胞、巨噬细胞、各种B细胞和T细胞等,在IgG4-RD中均被广泛激活[9]。同时有研究认为,B细胞、CD4+ CTL、Th2和调节性T细胞(regulatory T cell, Treg)均在发病过程中起到重要作用[10-15]。研究者在对IgG4-RD的治疗中,发现B细胞清除疗法具有很好的效果,进而推测B细胞在致病过程中起到了关键性作用。大量证据表明,Th2和Treg确实参与了IgG4-RD的病理生理过程[13-15]。在病变组织中发现了大量T细胞尤其是CD4+ CTL浸润。一项针对9例IgG4相关泪腺肿大患者的临床特征分析结果表明,患者CD4+ CTL的IL-4、IL-5、IL-10和GATA-3 mRNA表达较健康对照组增高[15]。有研究表明,Th2细胞因子和由Treg产生的IL-10参与调节B细胞表达IgG4免疫球蛋白的类别转换过程[16]。另一项通过流式细胞术鉴定IgG4-RD患者的T细胞的研究表明,IgG4-RD患者中IgG4+浆细胞是以T细胞依赖的方式产生的。此外,当从优势质母细胞中克隆成对的重链和轻链时,重组单克隆抗体具有自身反应性,可能导致人喉表皮样癌细胞(human laryngeal epidermoid, Hep-2)的明亮细胞质染色[17]。该研究首次证明了IgG4-RD的体液免疫反应是针对自身抗原的,并为如何识别这些抗原提供了线索。以上种种证据都支持IgG4-RD属于自身免疫病这一观点。而IgG4-ROD是IgG4-RD在眼部的疾病表现,同样可以认为IgG4-ROD属于自身免疫病。因此,在对IgG4-ROD疾病的认识、诊断和治疗等方面的研究可以通过与其他自身免疫性疾病相关联来获得新思路。3 IgG4-ROD与纤维化
IgG4-ROD多数具有纤维化的病理改变。纤维化是机体对抗疾病损伤的一种自我保护的组织改变,但失调时则会对机体造成不可逆的损伤,从而对疾病预后造成严重的负面影响。3.1 B细胞在IgG4-ROD纤维化进程中的作用
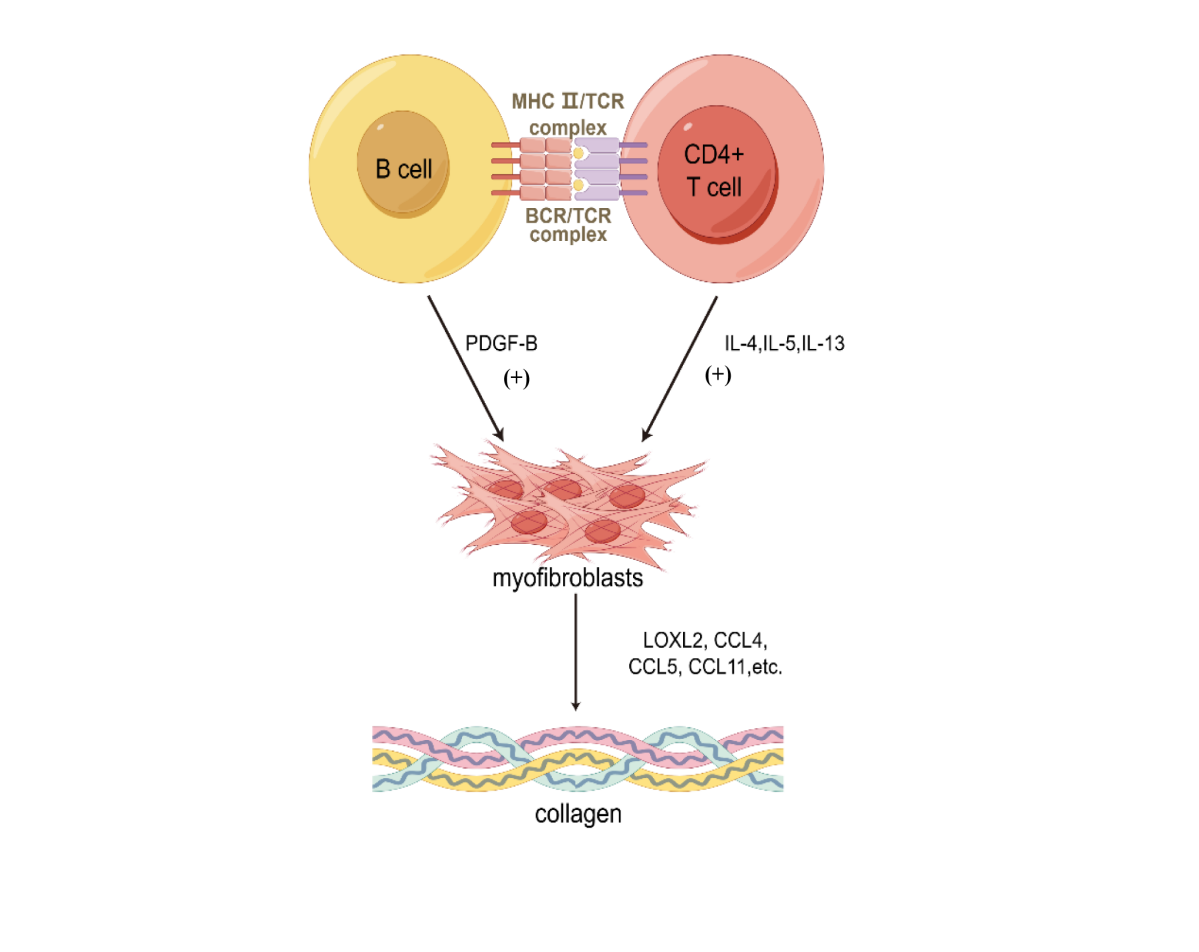
在IgG4-RD被认为是一种疾病实体以后,体液免疫在研究其发病机制时受到了广泛关注。Mattoo等[18]的一项研究表明,浆母细胞和其他活化的B细胞在病变组织中高度浸润。一方面,这些活化的B细胞通过表达主要组织相容性复合体2类分子(major histocompatibility complex 2, MHC II)MHC Ⅱ类分子及高亲和力的B细胞抗原受体(B-cell receptor, BCR),捕获自身抗原并在合适的MHCⅡ类分子加工,进而呈递给可能导致疾病的CD4+T细胞。另一方面,活化的B细胞直接释放细胞因子、趋化因子、生长因子、促炎蛋白及促纤维化蛋白,直接作用于成纤维细胞及作用于T细胞。这些分泌促炎蛋白和促纤维化蛋白的B细胞来源于表达CD20的B细胞。且大多数患者采用CD20 B细胞耗竭疗法可获得显著临床反应。另有一项研究通过人成纤维细胞与IgG4-RD患者的总循环CD19 B细胞、naive B细胞、记忆B细胞或浆母细胞的共培养,发现来自IgG4-RD患者的B细胞产生血小板衍生生长因子B(platelet derived growth factor B, PDGF-B)刺激成纤维细胞产生胶原,产生与细胞外基质重塑有关的赖酰胺氧化酶样蛋白2(lysyl oxidase like 2, LOXL2)及趋化因子CC 基序趋化因子配体 4(C-C motif chemokine ligand 4, CCL-4)、CC 基序趋化因子配体5(C-C motif chemokine ligand 5, CCL-5)和CC 基序趋化因子配体 11(C-C motif chemokine ligand 11, CCL-11),进而促进组织纤维化[10]。这些证据可能潜在地反映了活化的B细胞可能是纤维化的关键细胞驱动因素,有理由推测B细胞既支持CD4+CTL的活性,又分泌抗体以外的蛋白质,参与IgG4-RD纤维化进程。作为IgG4-RD在眼部的病变,同样有理由认为B细胞参与了IgG4-ROD的纤维化机制。3.2 T细胞在IgG4-ROD纤维化进程中的作用
CD4+T细胞在IgG4-RD纤维化发病机制中发挥了主要作用,主要包括 CD4+ Th2 、CD4+ Treg 、CD4+滤泡辅助性 T 细胞(follicular helper T cells, Tfh)与 CD4+ CTL [11-15,19]。研究者发现IgG4相关泪腺肿大患者外周血CD4+ T细胞的Th2分化和IL-10产生增强,表明Th2可能在IgG4-ROD的发病机制中起作用[15]。Heeringa等[12]的研究观察到IgG4-RD患者中Th2和Treg的数量增加,并且Th2与IgG4+ B细胞的数量呈正相关,证实Th2和Treg分泌IL-4和IL-10介导IgG4分子类转换。Th2细胞因子被认为具有很强的促纤维化能力,其中IL-4、IL-5 和 IL-13 与纤维化的发展有关。研究表明,成纤维细胞在被IL-4激活后会增加胶原蛋白和其他基质蛋白的合成,还可直接激活B细胞,诱导初始CD4+ T细胞向Th2 表型分化,并支持M2型单核细胞/巨噬细胞的发育。然而,IL-4还具有重要的免疫抑制特性,可以抑制炎性反应,从而在某些条件下可能促进纤维化的发展[20]。IL-4是启动Th2免疫反应的关键细胞因子,而IL-13在维持Th2免疫反应中发挥重要作用。IL-13还可以直接或间接激活成纤维细胞参与纤维化过程[21]。IL-13主要作用于IL-13Ra和IL-13Ra2两种受体促纤维化。IL-13或IL-4与TNF-α共同诱导巨噬细胞上IL-13Ra2的上调,而后受体结合IL-13,进一步诱导释放促纤维化因子转化生长因子β(transforming growth factor-β, TGF-β)。而IL-13Ra2被认为对IL-13的亲和力比IL-13Ra高1 000倍以上且同样具有促纤维化特性[22]。同时,IL-5同样被认为具有促纤维化作用[23]。不同的T细胞亚群可能与受累组织中的B细胞亚群相互作用,从而进一步导致下游的纤维生成。CD4⁺ CTL和活化的B细胞分泌趋化因子、细胞因子及其他促纤维化分子,这些分子可能诱导单核细胞、纤维细胞、B细胞和T细胞的招募,以及巨噬细胞、成纤维细胞和肌成纤维细胞的激活[9]。Maehara等[14]探究IgG4相关泪腺炎和涎腺炎患者CTL的作用的结果表明,患者样本中颗粒酶A (granzyme A, GZMA) mRNA的表达水平明显高于干燥综合征和健康对照组的相应组织。定量影像学显示,IgG4相关泪腺炎和涎腺炎患者的浸润性CD4+ GZMA+ CTL较其他组更为丰富,且下颌下腺中大部分CD4+ GZMA+ CTL分泌干扰素-γ。Mattoo等[17]发现,IgG4-RD组织中CD4+ SLAMF7+ CTL表达IL-1β和TGF-β1。IL-1β的过表达可能导致纤维化[24-25]。由CTL分泌的成纤维细胞生长因子结合蛋白2 (fibroblast growth factor-binding protein 2, FGFBP2)可与成纤维细胞生长因子结合,促进成纤维细胞合成胶原纤维[26]。上述研究均表明,CD4+ CTL可能参与诱导IgG4-RD中出现纤维化的关键环节。Tfh的特征是表达谱系特异性转录因子Bcl6和ICOS及分泌IL-21,对B细胞功能至关重要。同时Tfh还表达高水平的表面标志物,如CXC 基序趋化因子受体 5(C-X-C motif chemokine receptor 5, CXCR5)、CD40配体(CD40 ligand,CD40L)、诱导性共刺激分子(inducible costimulator,ICOS)和程序性死亡受体 1(programmed death receptor 1, PD-1)[27]。IgG4+浆细胞被认为可能来源于异位生发中心,在Tfh的帮助下大量扩增。组织学分析发现,在IgG4-RD患者受影响的唾液腺中观察到了异位生发中心[28]。Tfh需要上调Bcl-6来发育和发挥其在生发中心中B细胞成熟中的功能[29]。与此同时,B细胞在维持Bcl-6表达方面,也就是说在向Tfh分化的后期起到了关键作用[30]。这似乎形成了一个促进纤维化的循环。对此,Poholek等[29]提出了一个模型,认为CD4+ T细胞在T细胞区激活后上调Bcl6水平,从而抑制P选择素糖蛋白配体1(P-selectin glycoprotein ligand 1, PSGL1)的表达。PSGL1表达的减少使T细胞失去结合CCL19和CCL21的能力,从而去除了T区的保留信号,同时减少了CCR7的表达,这使得Bcl-6+ PSGL1低表达细胞能够向B细胞滤泡迁移。在T-B细胞边界,来自B细胞的额外信号通过促进Bcl6水平的升高以及PD-1和CXCR5的上调,促进了Tfh的分化和扩增。CXCR5的上调使Tfh能够进入B细胞滤泡,并通过CD40L、IL-21和其他激活信号促进生发中心中B细胞的存活和成熟。
图 1 B细胞和T细胞诱导IgG-ROD组织纤维化途径。
Figure 1 The pathways through which B cells and T cells induce IgG-ROD tissue fibrosis.
本图由Figdraw(http://www.figdraw.com)绘制。
The figurewas drawn by Figdraw (www.figdraw.com)
The figurewas drawn by Figdraw (www.figdraw.com)
4 IgG4-ROD从炎症到纤维化
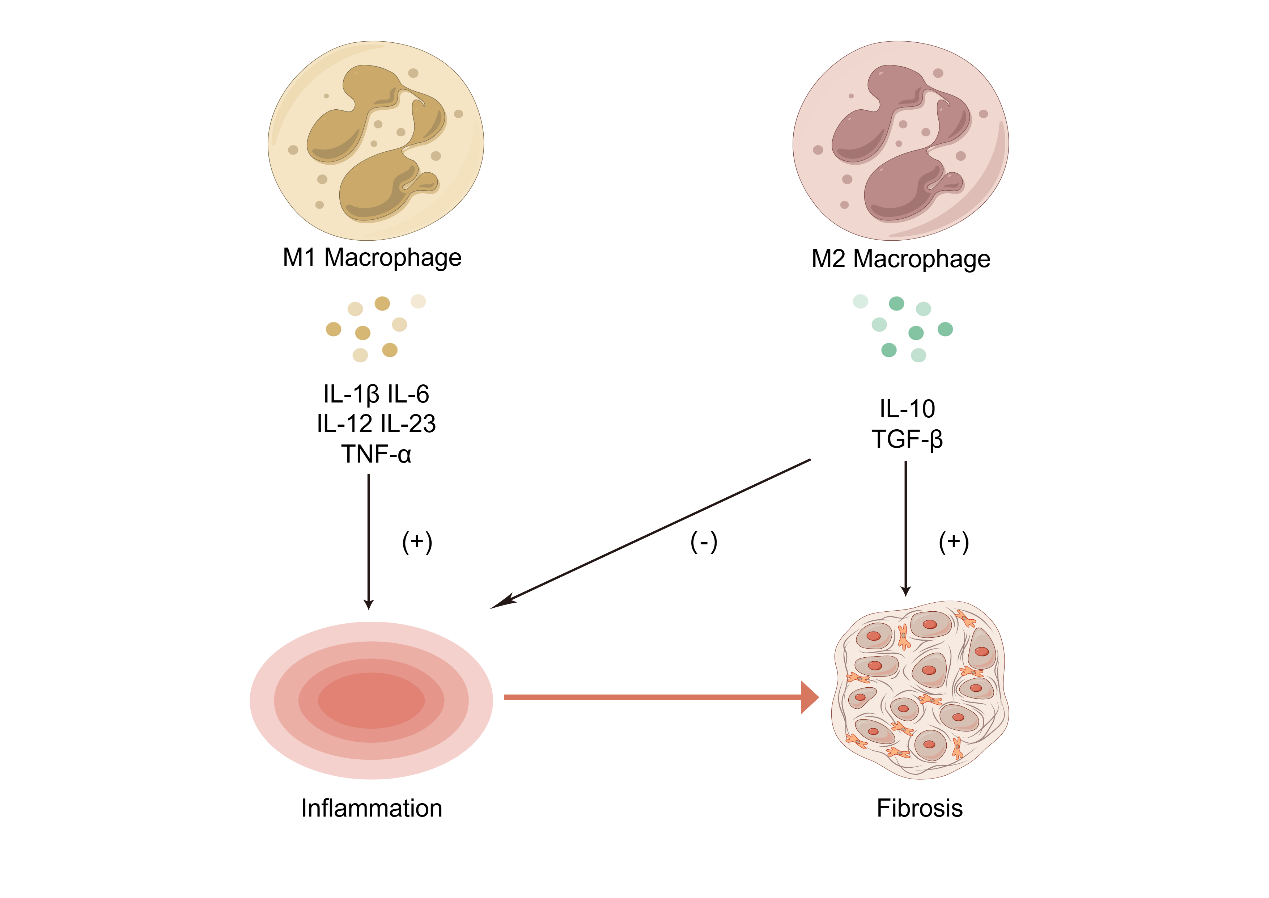
日本学者首先发现IgG4与Mikulicz病和硬化性泪腺炎存在联系[31-32]。该发现首次将IgG4-ROD从特发性眼眶炎症(idiopathic orbital inflammation, IOI)中作为一个疾病实体分离出来。McCarthy等[33]发现IOI的组织形态学和免疫组织化学与腹膜后纤维化之间存在相似性。IgG4-ROD的病理变化可能是一个动态的,从炎症向纤维化转变的过程。炎症反应可以刺激免疫细胞分泌促纤维化的趋化因子和细胞因子作用于成纤维细胞。炎性小体是由多种蛋白质组成的细胞内复合体,是先天免疫系统的重要组成部分。许多证据表明,炎性小体参与各种器官纤维化的发生[34-36]。因此,阐明炎性小体致组织纤维化的过程对于确定治疗纤维化的新治疗靶点是十分必要的。一项间隔7年、对同一IgG4-ROD患者的泪腺组织进行两次病理活检的结果显示,组织发生了从显著的滤泡淋巴浆细胞浸润到致密的胶原性纤维化的改变[37]。研究结果提示,这一动态变化过程可能是确切存在的,即免疫细胞异常激活,造成炎症浸润,分泌的炎性因子等物质驱动成纤维细胞活化、成纤维细胞募集、胶原蛋白沉积及细胞外基质形成。几种细胞及细胞因子在炎症过程和纤维化进展中发挥着重要作用。首先被发现的具有强烈促纤维化特性的是Th2及Th2细胞因子,其中与纤维化相关的细胞因子包括IL-4、IL-5和IL-13。有趣的是,IL-4在炎症与纤维化进程中的作用表现出矛盾的结果。IL-4很早就被发现作用于成纤维细胞的促纤维化[38]。然而,IL-4也具有重要的免疫抑制特性,可以阻断各种促炎介质,如IL-1、TNF-α、IL-6、IL-12和IL-8的表达。研究者在肺纤维化和肾脏纤维化的模型中都发现了IL-4减弱炎症的作用[39-40]。因此,IL-4在某些条件下对纤维化发展可能产生促进效应。无独有偶,IL-13也有类似的矛盾的特点,在直接或间接作用于成纤维细胞促纤维化的同时还具有抗炎特性[41]。巨噬细胞存在M1和M2两个亚群[42]。M1型巨噬细胞产生促炎细胞因子如IL-1β、IL-6、IL-12、IL-23和TNF-α,具有促炎作用;M2型巨噬细胞产生IL-10、TGF-β等抗炎细胞因子,具有抗炎和免疫调节作用。当感染或炎症初期,巨噬细胞首先表现出M1表型,即促进炎症的进展。当炎症进展到一定阶段,M2巨噬细胞增加来抑制炎症,参与组织修复、重塑、血管生成和维持体内平衡。其中M2巨噬细胞分泌的TGF-β是既具有抗炎作用又具有促纤维化功能的细胞因子。这也印证了在IgG4-ROD的炎症和纤维化两个阶段之间似乎存在某一个平衡点或交叉点。近年来的研究表明,一些非肽介质如活性氧(reactive oxygen species, ROS)、脂质介质和乙醛等,诱导与纤维发生相关的介质的表达或活性。过量的ROS产生、加速的脂质积累和乙醛的形成被认为是纤维化发病机制的关键因素。ROS的一些生物活性与TGF-β基因表达或活性的诱导直接相关,而其他生物活性似乎并不依赖TGF-β介导[43]。这些都提示我们应将IgG4-ROD的炎性反应与纤维化改变的分子机制联系起来,将会得到更加完整且严谨的结果。
图 2 M1和M2巨噬细胞对从炎症到纤维化动态转变的调控。
Figure 2 Regulation of the dynamic transition from inflammation to fibrosis by M1 and M2 macrophages.
本图由Figdraw(http://www.figdraw.com)绘制。
The figurewas drawn by Figdraw (www.figdraw.com).
The figurewas drawn by Figdraw (www.figdraw.com).
5 总结与展望
近年来,IgG4-ROD被确定为一个新的疾病实体,有关研究正如火如荼地开展。然而,对其致病分子机制的理解仍处于初步阶段。首先,需要明确IgG4在疾病中起到什么样的作用。其次,有关IgG4-ROD的纤维化的病理机制研究相对匮乏,如免疫细胞和成纤维细胞之间相互作用的具体机制等。最后,炎症与纤维化的动态变化过程仍有待进一步阐明,阻断有关信号通路可能是预防IgG4-ROD严重不良结局的关键。现有的治疗方案无论是单独使用糖皮质激素大剂量冲击还是单独使用免疫抑制剂抑或二者并用都无法做到完全治愈,且存在一定概率会复发。对IgG4-ROD的发病机制进行分子层面的探索,无疑将为精准治疗打下基础。总之,从分子视角对IgG4-ROD的发病机制进行研究将有助于我们更加了解该疾病,进而提出新的靶向分子治疗方案,为达到更好的疾病预后打下坚实基础。声明
在论文撰写中无使用生成式人工智能。论文撰写中的所有内容均由作者独立完成,并对出版物的真实性和准确性承担全部责任。利益冲突
所有作者均声明不存在利益冲突。开放获取声明
本文适用于知识共享许可协议(Creative Commons),允许第三方用户按照署名(BY)-非商业性使用(NC)-禁止演绎(ND)(CC BY-NC-ND)的方式共享,即允许第三方对本刊发表的文章进行复制、发行、展览、表演、放映、广播或通过信息网络向公众传播,但在这些过程中必须保留作者署名、仅限于非商业性目的、不得进行演绎创作。基金
暂无基金信息
参考文献
1. Kamisawa T, Zen Y, Pillai S, et al. IgG4-related disease[J]. Lancet, 2015, 385(9976): 1460-1471. DOI:10.1016/S0140-6736(14)60720-0.
2. Stone JH, Khosroshahi A, Deshpande V, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations[J]. Arthritis Rheum, 2012, 64(10): 3061-3067. DOI:10.1002/art.34593.
3. Umehara H, Okazaki K, Masaki Y, et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details[J]. Mod Rheumatol, 2012, 22(1): 1-14. DOI:10.1007/s10165-011-0508-6.
4. 中华医学会眼科学分会眼整形眼眶病学组, 中华医学会风湿病学分会. IgG4相关眼病诊治专家共识(2024版)[J]. 中华医学杂志, 2024, 104(40): 3726-3735. DOI:10.3760/cma.j.cn112137-20240814-01870.
Oculoplastic and Orbital Disease Group of Chinese Ophthalmological Society of Chinese Medical Association, Chinese Rheumatology Association. Expert consensus on the diagnosis and treatment of immunoglobulin-G4 related ophthalmic disease (2024 edition)[J]. Natl Med J China, 2024, 104(40): 3726-3735. DOI:10.3760/cma.j.cn112137-20240814-01870.
Oculoplastic and Orbital Disease Group of Chinese Ophthalmological Society of Chinese Medical Association, Chinese Rheumatology Association. Expert consensus on the diagnosis and treatment of immunoglobulin-G4 related ophthalmic disease (2024 edition)[J]. Natl Med J China, 2024, 104(40): 3726-3735. DOI:10.3760/cma.j.cn112137-20240814-01870.
5. Zen Y, Nakanuma Y. Pathogenesis of IgG4-related disease[J]. Curr Opin Rheumatol, 2011, 23(1): 114-118. DOI:10.1097/BOR.0b013e3283412f4a.
6. Rose NR. Autoimmune diseases: tracing the shared threads[J]. Hosp Pract, 1997, 32(4): 147-154. DOI:10.1080/21548331.1997.11443469.
7. Sun H, Zeng X, Li Y, et al. Successful remission induction of IgG4-related ophthalmic disease by obinutuzumab therapy: a retrospective study of 8 patients[J]. Eye, 2024, 38(4): 723-729. DOI:10.1038/s41433-023-02758-8.
8. Wu A, Andrew NH, Tsirbas A, et al. Rituximab for the treatment of IgG4-related orbital disease: experience from five cases[J]. Eye, 2015, 29(1): 122-128. DOI:10.1038/eye.2014.251.
9. Lu C, Li S, Qing P, et al. Single-cell transcriptome analysis and protein profiling reveal broad immune system activation in IgG4-related disease[J]. JCI Insight, 2023, 8(17): e167602. DOI:10.1172/jci.insight.167602.
10. Della-Torre E, Rigamonti E, Perugino C, et al. B lymphocytes directly contribute to tissue fibrosis in patients with IgG(4)-related disease[J]. J Allergy Clin Immunol, 2020, 145(3): 968-981.e14. DOI:10.1016/j.jaci.2019.07.004.
11. Perugino CA, Kaneko N, Maehara T, et al. CD4+ and CD8+ cytotoxic T lymphocytes may induce mesenchymal cell apoptosis in IgG(4)-related disease[J]. J Allergy Clin Immunol, 2021, 147(1): 368-382. DOI:10.1016/j.jaci.2020.05.022.
12. Heeringa JJ, Karim AF, van Laar JAM, et al. Expansion of blood IgG(4)(+) B, T(H)2, and regulatory T cells in patients with IgG(4)-related disease[J]. J Allergy Clin Immunol, 2018, 141(5): 1831-1843.e10. DOI:10.1016/j.jaci.2017.07.024.
13. Yang H, Wei R, Liu Q, et al. Frequency and distribution of CD4+CXCR5+ follicular B helper T cells within involved tissues in IgG4-related ophthalmic disease[J]. Mol Med Rep, 2017, 16(6): 9512-9520. DOI:10.3892/mmr.2017.7780.
14. Maehara T, Mattoo H, Ohta M, et al. Lesional CD4+ IFN-γ+ cytotoxic T lymphocytes in IgG4-related dacryoadenitis and sialoadenitis[J]. Ann Rheum Dis, 2017, 76(2): 377-385. DOI:10.1136/annrheumdis-2016-209139.
15. Kanari H, Kagami SI, Kashiwakuma D, et al. Role of Th2 cells in IgG4-related lacrimal gland enlargement[J]. Int Arch Allergy Immunol, 2010, 152(Suppl 1): 47-53. DOI:10.1159/000312125.
16. Tsuboi H, Matsuo N, Iizuka M, et al. Analysis of IgG4 class switch-related molecules in IgG4-related disease[J]. Arthritis Res Ther, 2012, 14(4): R171. DOI:10.1186/ar3924.
17. Mattoo H, Mahajan VS, Maehara T, et al. Clonal expansion of CD4+ cytotoxic T lymphocytes in patients with IgG4-related disease[J]. J Allergy Clin Immunol, 2016, 138(3): 825-838. DOI:10.1016/j.jaci.2015.12.1330.
18. Mattoo H, Mahajan VS, Della-Torre E, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease[J]. J Allergy Clin Immunol, 2014, 134(3): 679-687. DOI:10.1016/j.jaci.2014.03.034.
19. Munemura R, Maehara T, Murakami Y, et al. Distinct disease-specific Tfh cell populations in 2 different fibrotic diseases: IgG(4)-related disease and Kimura disease[J]. J Allergy Clin Immunol, 2022, 150(2): 440-455.e17. DOI:10.1016/j.jaci.2022.03.034.
20. Hart PH, Vitti GF, Burgess DR, et al. Potential antiinflammatory effects of interleukin 4: suppression of human monocyte tumor necrosis factor alpha, interleukin 1, and prostaglandin E2[J]. Proc Natl Acad Sci USA, 1989, 86(10): 3803-3807. DOI:10.1073/pnas.86.10.3803.
21. Roeb E. Interleukin-13 (IL-13)-a pleiotropic cytokine involved in wound healing and fibrosis[J]. Int J Mol Sci, 2023, 24(16): 12884. DOI:10.3390/ijms241612884.
22. Lumsden RV, Worrell JC, Boylan D, et al. Modulation of pulmonary fibrosis by IL-13Rα2[J]. Am J Physiol Lung Cell Mol Physiol, 2015, 308(7): L710-L718. DOI:10.1152/ajplung.00120.2014.
23. Gieseck RL 3rd, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis[J]. Nat Rev Immunol, 2018, 18(1): 62-76. DOI:10.1038/nri.2017.90.
24. Zhang WJ, Chen SJ, Zhou SC, et al. Inflammasomes and fibrosis[J]. Front Immunol, 2021, 12: 643149. DOI:10.3389/fimmu.2021.643149.
25. Li L, Xiang T, Guo J, et al. Inhibition of ACSS2-mediated histone crotonylation alleviates kidney fibrosis via IL-1β-dependent macrophage activation and tubular cell senescence[J]. Nat Commun, 2024, 15(1): 3200. DOI:10.1038/s41467-024-47315-3.
26. Abuharbeid S, Czubayko F, Aigner A. The fibroblast growth factor-binding protein FGF-BP[J]. Int J Biochem Cell Biol, 2006, 38(9): 1463-1468. DOI:10.1016/j.biocel.2005.10.017.
27. Liu X, Nurieva RI, Dong C. Transcriptional regulation of follicular T-helper (Tfh) cells[J]. Immunol Rev, 2013, 252(1): 139-145. DOI:10.1111/imr.12040.
28. Maehara T, Moriyama M, Nakashima H, et al. Interleukin-21 contributes to germinal centre formation and immunoglobulin G4 production in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease[J]. Ann Rheum Dis, 2012, 71(12): 2011-2019. DOI:10.1136/annrheumdis-2012-201477.
29. Poholek AC, Hansen K, Hernandez SG, et al. In vivo regulation of Bcl6 and T follicular helper cell development[J]. J Immunol, 2010, 185(1): 313-326. DOI:10.4049/jimmunol.0904023.
30. Beerli P, Palczewski M. Unified framework to evaluate panmixia and migration direction among multiple sampling locations[J]. Genetics, 2010, 185(1): 313-326. DOI:10.1534/genetics.109.112532.
31. Yamamoto M, Ohara M, Suzuki C, et al. Elevated IgG4 concentrations in serum of patients with Mikulicz’s disease[J]. Scand J Rheumatol, 2004, 33(6): 432-433. DOI:10.1080/03009740410006439.
32. Takahira M, Kawano M, Zen Y, et al. IgG4-related chronic sclerosing dacryoadenitis[J]. Arch Ophthalmol, 2007, 125(11): 1575-1578. DOI:10.1001/archopht.125.11.1575.
33. McCarthy JM, White VA, Harris G, et al. Idiopathic sclerosing inflammation of the orbit: immunohistologic analysis and comparison with retroperitoneal fibrosis[J]. Mod Pathol, 1993, 6(5): 581-587.
34. Hu Z, Chai J. Structural mechanisms in NLR inflammasome assembly and signaling[M]//Inflammasome Signaling and Bacterial Infections. Cham: Springer International Publishing, 2016: 23-42. DOI:10.1007/978-3-319-41171-2_2.
35. Lorenz G, Darisipudi MN, Anders HJ. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis[J]. Nephrol Dial Transplant, 2014, 29(1): 41-48. DOI:10.1093/ndt/gft332.
36. DeSantis DA, Ko CW, Wang L, et al. Constitutive activation of the Nlrc4 inflammasome prevents hepatic fibrosis and promotes hepatic regeneration after partial hepatectomy[J]. Mediators Inflamm, 2015, 2015: 909827. DOI:10.1155/2015/909827.
37. Andrew N, Kearney D, Selva D. IgG4-related orbital disease: a meta-analysis and review[J]. Acta Ophthalmol, 2013, 91(8): 694-700. DOI:10.1111/j.1755-3768.2012.02526.x.
38. Fertin C, Nicolas JF, Gillery P, et al. Interleukin-4 stimulates collagen synthesis by normal and scleroderma fibroblasts in dermal equivalents[J]. Cell Mol Biol, 1991, 37(8): 823-829.
39. Izbicki G, Or R, Christensen TG, et al. Bleomycin-induced lung fibrosis in IL-4-overexpressing and knockout mice[J]. Am J Physiol Lung Cell Mol Physiol, 2002, 283(5): L1110-L1116. DOI:10.1152/ajplung.00107.2002.
40. Liang H, Zhang Z, Yan J, et al. The IL-4 receptor α has a critical role in bone marrow-derived fibroblast activation and renal fibrosis[J]. Kidney Int, 2017, 92(6): 1433-1443. DOI:10.1016/j.kint.2017.04.021.
41. Mack M. Inflammation and fibrosis[J]. Matrix Biol, 2018, 68: 106-121. DOI:10.1016/j.matbio.2017.11.010.
42. Shapouri-Moghaddam A, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization, and function in health and disease[J]. J Cell Physiol, 2018, 233(9): 6425-6440. DOI:10.1002/jcp.26429.
43. Weiskirchen R, Weiskirchen S, Tacke F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications[J]. Mol Aspects Med, 2019, 65: 2-15. DOI:10.1016/j.mam.2018.06.003.