蜡样芽孢杆菌性眼内炎致病机制及研究进展
'%20fill='white'%20fill-opacity='0.01'/%3e%3cmask%20id='mask0_3477_29692'%20style='mask-type:luminance'%20maskUnits='userSpaceOnUse'%20x='0'%20y='0'%20width='16'%20height='16'%3e%3crect%20id='&%23232;&%23146;&%23153;&%23231;&%23137;&%23136;_2'%20x='16'%20width='16'%20height='16'%20transform='rotate(90%2016%200)'%20fill='white'/%3e%3c/mask%3e%3cg%20mask='url(%23mask0_3477_29692)'%3e%3cpath%20id='&%23232;&%23183;&%23175;&%23229;&%23190;&%23132;'%20d='M14%205L8%2011L2%205'%20stroke='%23333333'%20stroke-width='1.5'%20stroke-linecap='round'%20stroke-linejoin='round'/%3e%3c/g%3e%3c/g%3e%3c/svg%3e)
关键词
摘要
全文
文章亮点
1. 关键发现
2. 已知与发现
3. 意义与改变
在眼科急诊中,炎症和创伤是常见且重要的临床问题。它们不仅是患者就诊的主要原因,还可能导致视力受损,甚至永久性失明。随着生活方式的变化,眼科急诊的接诊量逐年上升,因此,及时诊断和治疗对于减轻眼科炎症导致的视力损害至关重要。眼内炎通常与眼外伤、手术、角膜感染扩展或眼内注射等因素相关。统计数据表明,眼外伤引起的眼内炎占所有病例20%~30%[1],这一比例在我国可高达58.5%[2]。在引发创伤性眼内炎的致病菌中,蜡样芽孢杆菌(bacillus cereus)是主要致病菌之一,仅次于表皮葡萄球菌[3]。
1 流行病学特征
1.1 形态学特征
蜡样芽孢杆菌是一种革兰阳性杆菌,具有周鞭毛,在高营养条件下可表现出超鞭毛化和细胞伸长,形成具有迁移特性的菌落[10-11]。其芽孢呈椭圆形,通常位于细胞中央或偏中心,芽孢形成不会导致细胞膨胀[10]。在普通琼脂培养基上,经37 ℃培养24 h,可形成具有蜡样光泽的圆形或近圆形菌落。
1.2 生长特性
蜡样芽孢杆菌的生长温度范围为 5~40 ℃,最适温度为 37 ℃。产呕吐毒素的菌株耐高温,腹泻型菌株则耐低温[11-13]。它能够在多种营养环境中生长,尤其是在富含碳源、乙醇和维生素 B1 的培养基中[14-15]。其适宜的 pH 范围为 4.5~7.5[16-19]。蜡样芽孢杆菌可形成耐热芽孢,在 100 ℃ 下可存活 0.6~27 min,并且在富含蛋白的环境中更易复苏[20-21]。它能够在土壤、食品和消化道中生存,在营养缺乏时通过形成芽孢提高存活率,在适宜条件下快速繁殖并形成生物膜[22-23]。
1.3 分布特点与传播途径
蜡样芽孢杆菌在自然环境中分布广泛,存在于土壤、食物、昆虫幼虫及人类皮肤等多种环境中[21]。其芽孢对热、干燥、辐射等极端环境条件具有很强的抵抗力,并通过各种途径传播到新的生态位[24]。蜡样芽孢杆菌凭借其广泛的分布和强大的生存能力,成为一种潜在的眼部感染源。文献指出,该菌引起的眼内炎主要与开放性眼外伤相关,尤其是当眼内存在异物(intraocular foreign body,IOFB)时,感染风险显著增加[25-27]。其芽孢可能通过被污染的物体(如金属碎片、植物碎片、玻璃碎片等)进入眼内,引发感染[25]。此外,在眼科手术或注射过程中,如果器械或操作环境未严格消毒,也可能成为感染源[28-29]。在某些病例中,尽管对使用的乳酸林格液、黏弹剂和超声乳化探头等进行了微生物学检测,未发现蜡样芽孢杆菌生长,但在手术室洗手区的水样中检测到与患者眼内样本相同的蜡样芽孢杆菌,提示手术室环境可能是感染的潜在来源[30]。
2 分子致病机制
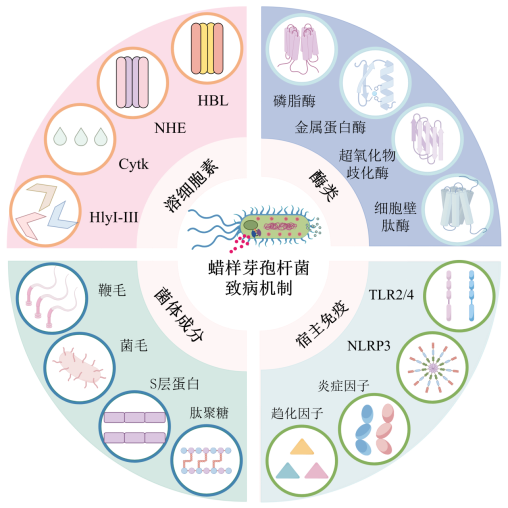
蜡样芽孢杆菌的分子致病机制十分复杂,主要可归纳为四大类:溶细胞素、多种酶类、菌体成分及其对宿主免疫的激活(详见图1)。
图1 蜡样芽孢杆菌眼内炎的主要致病机制
Figure 1 The main pathogenic mechanism of Bacillus cereus endophthalmitis
2.1 溶细胞毒素
蜡样芽孢杆菌在眼内炎病程中会分泌多种溶细胞毒素,主要包括溶血素BL(hemolysin BL,HBL)、非溶血性肠毒素NHE(non-hemolytic enterotoxin)、细胞毒素K(CytK,又名溶血素Ⅳ)、溶血素I、溶血素Ⅱ、溶血素Ⅲ等。这些溶细胞毒素通过破坏宿主细胞膜的完整性,诱导细胞死亡和炎症反应,从而在该菌的致病机制中发挥重要作用。2.1.1 成孔毒素
HBL和NHE是两种关键毒素复合物,通过激活NLR家族Pyrin结构域包含蛋白3(NLR family, pyrin domain containing 3, NLRP3)炎症小体发挥重要作用[31]。原核转录组分析发现,在眼内炎感染早期(8 h),HBL和NHE的表达量即可显著增加[32]。
HBL毒素通过B、L1和L2亚基依次组装形成跨膜孔道,诱导钾离子外流并激活NLRP3炎症小体,最终引发细胞焦亡[31, 33-34]。HBL的宿主受体包括脂多糖诱导的肿瘤坏死因子-α因子(LPS-inducible TNF-α factor, LITAF)和诱导细胞死亡的p53靶基因1(cell death-involved p53 target 1, CDIP1),其中LITAF是主要受体,CDIP1在LITAF缺失时作为替代受体[35]。HBL通过破坏视网膜色素上皮(retinal pigment epithelium, RPE)细胞和脉络膜毛细血管,增加血管通透性,引发炎症反应[36]。然而,缺乏 HBL 的蜡样芽孢杆菌分离株仍可致病,这表明其他非冗余毒力因子在该病原体的发病机制中也发挥着重要作用[37]。
研究发现NHE与HBL具有类似的致病机制,也依赖于亚基的逐步组装,但其成孔速度较慢且孔洞更大[38-39]。NHE 不仅对免疫细胞如小鼠骨髓来源的巨噬细胞(bone marrow-derived macrophage, BMDM)和人类单核细胞(tohoku hospital pediatrics-1,THP-1)具有细胞毒性,还能够影响多种非免疫细胞。这些细胞系在体外实验中表现出对NHE的敏感性[38]。尽管这些细胞类型并不自然存在于眼内,但在感染或炎症情况下,类似机制可能通过影响眼内固有细胞(如小胶质细胞和视网膜色素上皮细胞)或通过血流招募外周免疫细胞(巨噬细胞或中性粒细胞)来加剧眼内组织的损伤。
HBL在凝胶扩散测定中显示出独特的环形溶血型 (haemolytic type,HT)[40]。NHE最初被描述为没有溶血活性,因此被指定为“非溶血性”肠毒素。然而,最近也显示 NHE 对几种哺乳动物物种的红细胞具有溶血作用[41],且HBL和NHE在眼内转录水平显著升高[32],HBL和NHE可能是导致眼内炎咖啡样溶血的关键独立蛋白。
CytK是一种单一蛋白质、β桶形成孔毒素[42-44],作为一种重要的致病因子,其致病机制和作用逐渐受到关注。CytK通过与细胞质膜的相互作用结合巨噬细胞,触发由半胱氨酸酶1(caspase-1)激活的焦孔素D(gasdermin D,GSDMD)介导的快速焦亡[45]。研究发现,CytK对多种细胞膜脂质具有显著结合能力,尤其是磷脂酸,这种结合对CytK诱导的细胞死亡至关重要[45]。CytK与HBL/NHE受相同的群体感应系统plcR/papR调控[46],且都被相同的宿主受体识别,推测其可能同样作为一种非冗余毒素参与破坏眼部组织并加剧炎症反应。
2.1.2 溶血素
该菌不仅因HBL、NHE和CytK具备成孔特性而能诱导红细胞发生溶血现象,而且由于能表达溶血素I、溶血素Ⅱ和溶血素Ⅲ,从而表现出更广泛的溶血作用。
其中溶血素I(亦称溶血素O,CerO)是胆固醇依赖性溶血素(cholesterol-dependent cytolysins, CDCs)家族的成员,对红细胞表现出显著的溶血作用[40, 47]。CerO通过激活NLRP3炎症小体,诱导细胞焦亡,其C末端十一肽中的色氨酸残基对其膜结合及穿孔至关重要[48]。研究发现,CerO缺陷菌株在感染后4 h显著下调TLR4依赖的炎症介质,表明其可能通过TLR4激活炎症反应[32]。
溶血素Ⅱ(hemolysin II, HlyII)则属于分泌性β-桶孔形成毒素(β-barrel pore-forming toxin, β-PFT),能够形成七聚体孔道,导致细胞膜通透性增加,从而引发细胞凋亡[49-50]。HlyⅡ的表达受到特异性转录因子HlyⅡR和全局调节因子Fur的负向调控[51-53]。它能够诱导多种吞噬细胞(如昆虫血细胞、小鼠巨噬细胞和人单核细胞)的裂解,显示出广泛的细胞毒性作用[54]。此外,HlyⅡ的C末端片段(HlyⅡLCTD,Met225至Ile412)能够凝集兔红细胞,但不会导致红细胞裂解,这种凝集作用可能通过多种机制增强HlyII的致病能力,包括增强毒素的细胞结合能力、促进毒素的传播和扩散、激活免疫反应等[55]。
目前对溶血素Ⅲ(HlyⅢ)知之甚少,这种毒素尚未被纯化,甚至未明确它是否由蜡样芽孢杆菌组的成员分泌[40]。研究表明,HlyⅢ可能是一种孔形成型毒素能够通过多步骤机制破坏红细胞膜的完整性[56],且HlyⅢ的活性表现出明显的温度依赖性[56]。
综上所述,蜡样芽孢杆菌分泌的溶细胞素不仅通过破坏细胞膜完整性直接诱导细胞死亡,还通过激活炎症小体和炎症介质间接加剧炎症反应。未来研究需进一步探索这些溶细胞素在眼内炎中的具体机制及相互作用,为临床治疗提供依据。
2.2 酶类
2.2.1 磷脂酶蜡样芽孢杆菌分泌的膜损伤毒素包括鞘磷脂酶(sphingomyelinase, SMase)、磷脂酰胆碱特异性磷脂酶C(phosphatidylcholine-specific phospholipase C, PC-PLC)和磷脂酰肌醇特异性磷脂酶C(phosphatidylinositol-specific phospholipase C, PI-PLC)。
SMase是一种34 kDa的蛋白质,通过水解细胞膜中的鞘磷脂生成神经酰胺,改变膜的流动性和稳定性[57-58]。在巨噬细胞中,SMase降低膜流动性,抑制吞噬作用,帮助细菌逃避免疫清除[58];而在红细胞中,SMase则通过形成神经酰胺富集区域,导致膜的刚性与流动性区域分离,最终在物理刺激下引发膜的破坏和溶血[59]。另外,SMase还与其他毒素如NHE和HBL具有协同作用,显著增强其细胞毒性和溶血能力[60-61]。
PC-PLC表现出显著的体外毒性,能够引起视网膜细胞坏死[62]。有研究者发现,D609(三环葵烷-9-基-二硫代碳酸酯钾盐)作为PC-PLC的一种选择性抑制剂,能有效抑制视网膜色素上皮细胞(RPE细胞)的氧化损伤,从而可用于预防或治疗因氧化应激损伤RPE引起的视网膜损伤性疾病[63]。PI-PLC通过水解磷脂酰肌醇(phosphatidylinositol, PI)和PI-糖苷锚定蛋白,影响细胞膜的完整性和功能,从而干扰细胞的正常生理功能[64]。近期研究发现,PI-PLC蛋白会通过磷脂酰肌醇3-激酶(phosphoinositide 3-kinase, PI3K)/AKT(蛋白激酶B)信号通路抑制Müller细胞活力和吞噬活性,促进细菌的存活和繁殖[65]。
尽管目前的研究已经揭示了蜡样芽孢杆菌分泌的膜损伤毒素(如SMase、PC-PLC和PI-PLC)在眼内炎中的多种致病机制,但这些毒素在眼内微环境中的具体作用以及它们与其他宿主防御机制之间的相互作用仍有许多未知之处亟待探索。
2.2.2 金属蛋白酶
蜡样芽孢杆菌的两种金属蛋白酶InhA1和NprA被认为是重要的毒力因子[66]。在蜡样芽孢杆菌中,InhA1的表达水平显著高于其他金属蛋白酶[67, 68]。InhA1与芽孢外衣的结合使其在被巨噬细胞吞噬后仍能存活并逃逸,这一特性对于细菌在宿主体内的生存和传播至关重要[69]。
不仅如此,InhA1在特定位点切割NprA,使NprA的成熟形式(35 kDa)在野生菌株的上清中释放,该活性NprA形式对于蜡样芽孢杆菌逃逸巨噬细胞是必需的[70]。值得注意的是,NprA的表达受到nprR基因的调控,而该基因同时还影响细胞生存、孢子形成和抗菌药物抗性等多个基因的表达,从而显著影响细菌的毒力和适应性[32]。
在眼内炎的感染环境中,InhA1基因和nprR基因的表达水平均远高于BHI培养基[32]。这种上调表达提示金属蛋白酶在眼内炎的致病过程中可能扮演关键角色。
2.2.3 细胞壁肽酶
CwpFM(EntFM)是蜡样芽孢杆菌眼内炎中的潜在致病因子,其功能及作用机制日益受到关注。研究发现,蜡样芽孢杆菌的CwpFM参与细菌细胞壁的动态变化,显著影响细菌的运动性、黏附能力和生物膜形成[71-72]。这些特性对于细菌在复杂环境中的生存和致病性至关重要,尤其是在眼内这种免疫敏感且营养有限的环境中。
最近一项研究揭示了CwpFM在肠道感染中的致病机制。研究表明,蜡样芽孢杆菌分泌的CwpFM通过抑制核因子E2相关因子2(nuclear factor erythroid 2-related factor 2,NFE2L2) /血红素加氧酶-1(heme oxygenase-1,HO-1)信号通路,增加活性氧(reactive oxygen species, ROS)的积累,从而激活NLRP3炎症体和核因子κB (nuclear factor kappa B, NF-κB)炎症通路[73]。然而,研究人员通过RNA-Seq技术发现CwpFM在眼内炎小鼠模型中的表达水平与体外培养条件相比并无显著差异[32],考虑到RNA水平的表达并不一定完全反映蛋白质的实际含量或活性,因此CwpFM在感染过程中可能具有一定的功能,但其在眼内炎中的具体作用仍需进一步研究。
2.2.4 超氧化物歧化酶
超氧化物歧化酶(superoxide dismutase,SOD)作为一种关键的抗氧化酶,广泛存在于病原菌中,其主要功能在于保护细胞免受内源性ROS的损伤。在需氧及兼性厌氧菌的正常呼吸代谢过程中,超氧阴离子(O₂⁻)的产生不可避免,而SOD通过将其转化为过氧化氢(H₂O₂)和氧气(O₂),显著降低了这些活性氧对细胞的毒性[74-75]。在蜡样芽孢杆菌中,SOD同样发挥着重要作用,是其抗氧化防御系统的关键组成部分[76-77]。
研究表明,在体外玻璃体液环境中以及蜡样芽孢杆菌眼内炎的小鼠模型中,sodA2基因的表达水平均高于其他已知和假定的毒力因子[32, 78]。这一现象提示SOD可能在蜡样芽孢杆菌的眼内感染中扮演重要角色,帮助细菌抵御宿主中性粒细胞或自身产生的超氧阴离子,从而促进细菌的存活和感染的进展。
这一发现也为蜡样芽孢杆菌眼内炎的治疗策略提供了新的思路。若SOD在眼内炎的发生、发展中确实发挥关键作用,则开发针对SOD活性的抑制剂可能增强宿主的免疫防御能力,从而抑制细菌的感染。此外,已有研究指出外源性SOD能够调节宿主的氧化应激反应[79]。这表明通过调节宿主的抗氧化系统,例如使用抗氧化剂,可能有助于缓解蜡样芽孢杆菌眼内炎所引起的炎症反应和组织损伤。
2.3 菌体成分
2.3.1 鞭毛蜡样芽孢杆菌的鞭毛在眼内炎中的作用是多方面的,其不仅赋予细菌强大的运动能力,还与毒素分泌以及细菌在眼内环境中的适应性密切相关。研究表明,蜡样芽孢杆菌在眼内感染过程中表现出快速地生长和迁移能力,这种能力使其能够迅速扩散至角膜和其他眼部结构,导致严重的眼内炎和组织结构破坏[9, 80-81]。不仅如此,蜡样芽孢杆菌鞭毛的分泌装置可能通过与信号肽酶的协作促进HBL毒素的分泌[82],从而增强了蜡样芽孢杆菌在眼内感染中的致病性。
然而,与通常情况下Toll样受体5(toll-like receptor 5, TLR5)识别细菌鞭毛蛋白并激活宿主炎症反应的机制不同,蜡样芽孢杆菌的鞭毛在体外被证明是TLR5的弱激动剂[83]。这种特性可能使蜡样芽孢杆菌在宿主免疫系统中“隐形”,从而逃避有效的免疫清除。
有趣的是蜡样芽孢杆菌还表现出一种独特的丝状表面运动,这种运动不依赖鞭毛,但可能在眼内环境中表现出显著的适应性,尤其是在低营养、高黏度和微需氧的条件下[84]。这种丝状运动可能帮助细菌在眼内环境中更有效地扩散和逃避宿主免疫系统的攻击,从而增强其致病性。
2.3.2 菌毛
在蜡样芽孢杆菌眼内炎的致病机制中,菌毛的作用相对有限。在眼内环境中,野生型蜡样芽孢杆菌(ATCC 14579)表现出显著高于菌毛缺失突变株(ΔPil)的眼内生长能力,这提示菌毛可能在一定程度上促进细菌的存活和增殖[85-86]。然而,感染ΔPil或野生型蜡样芽孢杆菌的眼睛达到了相似程度的严重炎症,且视网膜损伤差异不显著,说明菌毛对视网膜功能的损害并非主要因素[85]。
2.3.3 S层蛋白
S层蛋白是一种覆盖在细菌细胞表面的有序晶体结构,广泛存在于多种细菌中[87-88]。在蜡样芽孢杆菌的不同菌株中,仅部分菌株能够表达S层蛋白,而这一特征可能与该细菌的致病性密切相关[88]。研究表明,缺乏S层蛋白的突变株与视网膜细胞(如Müller细胞、视网膜色素上皮细胞和光感受器细胞)的黏附能力显著降低,这表明S层蛋白可能通过增强细菌与宿主细胞的结合能力来促进感染发生[89-90]。此外,S层蛋白还能够激活视网膜Müller细胞中的NF-κB信号通路,进而促进炎症介质的表达[91]。在免疫逃逸方面,S层蛋白为蜡样芽孢杆菌提供了一种保护机制,使其能够免受Müller细胞和中性粒细胞的吞噬作用,从而逃避宿主免疫系统的清除[89]。除此之外,蜡样芽孢杆菌的S层蛋白还具有直接激活宿主免疫反应的能力。具体而言,S层蛋白能够激活TLR2和TLR4介导的信号通路,从而引发炎症反应[89]。
未来研究需进一步阐明S层蛋白的具体作用机制,以及如何通过靶向S层蛋白开发新的抗菌策略。
2.4 宿主免疫
2.4.1 Toll样受体蜡样芽孢杆菌眼内炎的发病机制不仅涉及细菌的直接毒性作用,还与宿主的免疫反应密切相关。TLR在识别病原体相关分子模式(pathogen-associated molecular pattern, PAMP)后,能够激活下游信号通路,促进中性粒细胞(polymorphonuclear neutrophil, PMN)和炎性因子的募集,从而在清除病原体方面发挥重要作用。然而,PMN和炎性因子在眼部感染中表现出明显的双刃剑效应:它们既有助于病原体的清除,也可能导致眼部组织损伤[92]。
TLR2和TLR4在蜡样芽孢杆菌眼内炎中对炎症反应的强度有显著影响。研究表明,TLR2和TLR4缺陷小鼠在感染后表现出较轻的眼内炎症,中性粒细胞浸润减少,促炎细胞因子和趋化因子水平降低,视网膜功能丧失的速度也显著减缓[93-95]。此外,TLR4可能通过其非经典配体(如溶血素O)的激活参与炎症反应[93]。
炎性因子如TNF-α,是PMN招募和炎症反应的关键介质,其在控制眼内病原体和维持视网膜功能方面发挥着至关重要的作用[96]。TNF-α在感染后2 h即可被检测到,并在10 h达到峰值,而PMN是感染后早期浸润的主要细胞类型,其浸润与TNF-α的增加同步[97]。然而,PMN在发挥抗菌作用的同时所释放的活性氧、蛋白酶和细胞外陷阱等产物则会对视网膜细胞和组织造成严重损伤[92]。
此外,趋化因子如C-X-C模体趋化因子配体1(C-X-C motif chemokine ligand 1, CXCL1)、 CXCL2、C-C模体趋化因子配体2(C-C motif chemokine ligand 2, CCL2)和CCL3 也在炎症反应中发挥重要作用。CXCL1和CXCL2的缺失或阻断能够显著减轻炎症并保护视网膜功能,而CCL2和CCL3的缺失也显示出对视网膜功能的保护作用,尽管CCL3的保护效果随时间减弱[98-100]。上述结果进一步证实了炎性因子在眼内炎中的复杂作用。因此,PMN和炎性因子的两面性效应提示我们在治疗过程中需要精准调控炎症反应,以实现病原体清除与组织保护的平衡。
2.4.2 炎症小体
炎性小体是一种细胞质蛋白复合物,由传感器蛋白、衔接蛋白(如凋亡相关斑点蛋白)和半胱氨酸蛋白酶(如caspase-1)构成,传感器蛋白如NLRP1、NLRP3等可以识别病原体和危险信号。激活后,炎性小体促进IL-1β和IL-18切割分泌,并诱导gasdermin D分裂触发焦亡[101-102]。如前所述,蜡样芽孢杆菌在引发感染的过程中,该菌毒素HBL、NHE和CytK均能激活炎症小体,诱导焦亡发生。其中,HBL和NHE通过引起钾离子外流激活NLRP3炎症小体,而CytK激活NLRP3炎症小体的方式还包括细胞内钙离子的积累[31, 38, 45]。
3 治疗现状与研究进展
3.1 常规治疗方案
蜡样芽孢杆菌眼内炎确诊后需立即全身和局部用抗菌药物,该菌对不同抗菌药物敏感性有差异,首选万古霉素[81]。有研究认为蜡样芽孢杆菌感染后4 h是玻璃体内注射万古霉素以挽救有用视力的关键时间[4]。此外,短时间内的外科手术干预很重要,玻璃体切割术(pars plana vitrectomy, PPV)是治疗蜡样芽孢杆菌性眼内炎的重要方法[103],可改善视力、降低致残风险,但治疗窗口短[104]。
3.2 新型治疗方式研究进展
3.2.1 临床研究基于PPV的治疗策略包括:1)在常规PPV基础上采用内镜辅助;2)临床确诊后立即行PPV并联合玻璃体腔内注射抗菌药物。结果显示,10例常规PPV与5例内镜辅助PPV在术后视力及眼球摘除率方面差异无统计学意义,但内镜辅助组就诊前症状持续时间更长;另有资料表明,预后视力(visual acuity, VA)较好的患者均接受PPV,其比例高于视力较差者,且保留眼球治疗组较眼球摘除术(enucleation of the eye, ENEV)组更频繁实施PPV。由此推断,内镜辅助PPV可作为替代方案,尤其适用于症状持续>6 h的病例;同时,早期实施PPV并联合抗菌药物有助于视力恢复。研究局限在于内镜辅助可能导致症状持续时间延长并增加不适,且未充分考虑PPV技术进步对远期及潜在视力结局的影响(表1)。
表1 传统与内镜辅助玻璃体切割术治疗效果的临床对比研究
Table1 Comparative outcomes of conventional vs. endoscope-assisted vitrectomy
|
研究策略 |
研究结果 |
研究结论 |
研究缺陷 |
|
基于PPV手术的基础上,用内镜辅助PPV[4]
|
10例行传统PPV患者和5例内镜辅助患者术后视力、眼球摘除率差异无统计学意义,但内镜辅助组症状持续时间更长 |
可作为一种替代治疗方案,尤其适用于症状持续时间超过6 h的病例 |
术后症状持续时间延长,患者不适感强 |
|
临床确诊后立即进行PPV手术联合玻璃体内注射抗菌药物治疗[105] |
视力良好组患者均接受PPV,高于 VA 较差组(100% vs 48.8%)。同样,保留眼睛治疗组比 ENEV 组更频繁地进行 PPV 治疗 |
早期的PPV和抗菌药物治疗有利于预后的视力恢复 |
未考虑PPV技术不断改良可能存在的长期和潜在视力影响 |
在蜡样芽孢杆菌眼内炎小鼠模型中,免疫调节策略(抗 TNF-α 或阻断 CCL2/CCL3)可显著减少中性粒细胞浸润、降低眼内炎症并保留/改善视网膜功能;以噬菌体溶素 PlyB 为核心的抗菌策略(单用或与 TLR2/4 抑制剂 OxPAPC 联用)则通过快速杀菌使眼内细菌负荷降至 <50 cfu/眼,维持视网膜结构,减轻炎性细胞浸润,并提高 ERG A、B 波振幅保留率。总体而言,两类干预均在动物模型中显示出调控炎症与保护视网膜功能的潜在益处,但目前仍缺乏充分的临床验证,其安全性与有效性尚需进一步评估(表2)。
表2 蜡样芽孢杆菌性眼内炎的实验治疗进展:免疫靶向与噬菌体溶素
Table2 Emerging experimental therapies for Bacillus cereus endophthalmitis: immune-targeted approaches and phage-derived lysins
|
研究策略 |
研究结果 |
研究结论 |
研究缺陷 |
|
应用免疫调节药物治疗,通过阻断特定炎症介质的表达来减轻炎症[96, 100] |
蜡样芽孢杆菌感染的小鼠通过抗TNF-α治疗或抗CCL2和CCL3治疗可减少中性粒细胞浸润、降低眼内炎症,并有效保留视网膜功能 |
在小鼠中有效调节眼内炎症,改善视网膜功能 |
研究基于实验模型,尚未在临床中得到充分验证 |
|
基于噬菌体溶素PlyB和TLR2/4抑制剂(OxPAPC)等联合治疗蜡样芽孢杆菌性眼内炎[106] |
噬菌体溶素单独或联合治疗后小鼠眼内细菌载量减少(低于50 cfu/眼)、视网膜结构基本完好、炎症细胞浸润显著减少、A波和B波振幅保留率显著提高 |
通过快速杀死细菌,减轻炎症,有效保护视网膜结构和功能
|
临床实用性存在限制,治疗方案的安全性和有效性需要评估 |
4 总结与展望
蜡样芽孢杆菌性眼内炎是一种暴发性高致盲性感染,其致病机制涉及病原体与宿主的多维度互作。在致病机制层面,近年研究逐渐勾勒出蜡样芽孢杆菌的“攻击路线图”:以HBL、NHE为代表的成孔毒素,在靶细胞表面形成跨膜通道,引发钾离子外流和NLRP3炎症小体激活。这种膜攻击并非孤立事件,如HlyⅡ与SMase协同作用可加速红细胞溶解,因此单一毒素抑制无法有效缓解组织损伤。以上提示细菌存在多毒力因子代偿机制,需开发联合阻断策略。
在临床治疗策略方面,现阶段一线疗法仍以PPV联合广谱抗菌药物(如万古霉素+头孢他啶)为主,但受限于血-视网膜屏障药物渗透率及4 h黄金干预窗口期,亟待突破性治疗方案的开发。本综述通过系统生物学方法解析蜡样芽孢杆菌关键毒力因子(如HBL/NHE/cytk)与Toll样受体/NLRP3炎症小体的分子互作网络,据此提出小分子抑制剂和毒素中和抗体治疗策略。未来研究需构建多靶点协同治疗体系,例如将万古霉素/噬菌体溶素plyB与NLRP3抑制剂MCC950联用,同步实现病原体清除和炎症风暴抑制。
值得注意的是,当前部分基础研究采用的细胞和动物模型未能精准模拟眼内炎病理特征(如玻璃体咖啡渣样出血、中性粒细胞浸润等关键表型),导致分子机制研究与临床表型研究存在脱节现象。建议未来构建具有眼内炎典型病理特征的疾病模型,通过建立标准化的临床前研究体系,强化基础研究向临床转化的关键节点,这将有助于揭示眼内炎发病机制的本质规律。
综上所述,解析毒力因子代偿网络和优化靶向递送与免疫调节联合疗法,将为治疗蜡样芽孢杆菌眼内炎提供坚实的实验和理论基础。