铁死亡机制在糖尿病视网膜病变中的研究进展
'%20fill='white'%20fill-opacity='0.01'/%3e%3cmask%20id='mask0_3477_29692'%20style='mask-type:luminance'%20maskUnits='userSpaceOnUse'%20x='0'%20y='0'%20width='16'%20height='16'%3e%3crect%20id='&%23232;&%23146;&%23153;&%23231;&%23137;&%23136;_2'%20x='16'%20width='16'%20height='16'%20transform='rotate(90%2016%200)'%20fill='white'/%3e%3c/mask%3e%3cg%20mask='url(%23mask0_3477_29692)'%3e%3cpath%20id='&%23232;&%23183;&%23175;&%23229;&%23190;&%23132;'%20d='M14%205L8%2011L2%205'%20stroke='%23333333'%20stroke-width='1.5'%20stroke-linecap='round'%20stroke-linejoin='round'/%3e%3c/g%3e%3c/g%3e%3c/svg%3e)
关键词
摘要
铁死亡(ferroptosis)是一种铁依赖性的程序性细胞死亡方式,以脂质过氧化物(lipid peroxide, LPO)异常蓄积和氧化还原稳态失衡为特征,被证实参与眼底疾病及代谢性疾病的病理进程,其分子机制的探索已成为当前转化医学研究的热点领域。近年研究发现,糖尿病视网膜病变(diabetic retinopathy, DR)的发生、发展与铁死亡密切相关,其病理过程涉及铁代谢紊乱、抗氧化系统崩溃及脂质过氧化级联反应。本文综述了铁死亡与DR发生、发展过程中的分子关联,重点阐明了高糖环境下视网膜细胞铁死亡的触发通路,进一步分析发现,铁死亡与线粒体功能障碍及炎症反应形成正反馈循环,通过氧化应激-炎症-血管新生轴加剧血-视网膜屏障破坏和神经血管单元损伤。本文论述了靶向铁死亡的干预策略的转化潜力,并提出未来需聚焦于阶段特异性生物标志物筛选和动态调控网络解析,为进一步探索DR的发病机制与防治手段提供新方向。
全文
文章亮点
1 关键发现
• 本文的关键发现在于证实铁死亡与糖尿病视网膜病变(diabetic retinopathy, DR) 的发生、发展密切相关,其病理过程涉及铁代谢紊乱、抗氧化系统崩溃及脂质过氧化级联反应。
2 已知与发现
• 已知铁死亡是一种铁依赖性的程序性细胞死亡方式,以脂质过氧化物蓄积和谷胱甘肽过氧化酶4抗氧化系统失调为特征,既往研究证实其参与肿瘤、神经退行性疾病等病理过程。
• 本文发现在DR中,铁死亡的具体调控路径得以明确: 高糖通过激活血管紧张素Ⅱ和转铁蛋白受体,显著增强视网膜细胞对铁的摄取能力,导致细胞内铁超载。视网膜微血管内皮细胞和光感受器因富含多不饱和脂肪酸,对铁死亡具有特殊易感性, 长链脂酰辅酶A合成酶4异常激活是核心事件。 线粒体功能障碍与铁死亡交互作用,硫氧还蛋白相互作用蛋白诱导线粒体功能障碍,导致受损线粒体堆积并触发铁释放。
3 意义与改变
• 本综述通过阐明铁死亡在DR中的分子关联,为进一步探索DR的发病机制与防治手段提供新方向。研究将铁代谢紊乱、脂质过氧化与DR的多种病理环节串联,构建了“氧化应激-炎症-血管新生轴”的综合模型。铁死亡诱导剂和抑制剂的开发推动了铁死亡靶向治疗从基础研究向临床转化迈进,为DR治疗提供新策略。未来需要进一步解析铁代谢动态平衡与氧化应激网络在疾病不同阶段的协同效应,并筛选特异性生物标志物以推动分层诊疗体系构建。
糖尿病视网膜病变(diabetic retinopathy, DR)是糖尿病最常见的微血管并发症之一[1],其病理机制涉及代谢紊乱、氧化应激、炎症反应、血管新生及神经退行性变等多重因素[2]。近年来,铁死亡(ferroptosis)作为一种新型铁依赖性细胞死亡方式,因其与氧化损伤的密切关联,逐渐成为DR研究的热点。
铁死亡是一种由抗氧化系统失调、铁代谢异常、脂质过氧化及p53通路激活等机制诱导的程序性细胞死亡形式[3]。其通过铁依赖的脂质过氧化损伤细胞膜结构,与肿瘤发展、神经退行性疾病、缺血再灌注损伤及器官纤维化等密切相关[4]。目前已开发出铁螯合剂、谷胱甘肽过氧化物酶4 (glutathione peroxidase 4, GPX4)抑制剂等治疗策略,在精准治疗和器官保护领域展现潜力。本文综述铁死亡的分子机制及其在DR中的研究进展,探讨其作为潜在治疗靶点的价值。
1 铁死亡的分子机制与特征
铁死亡作为铁依赖性的程序性细胞死亡方式,其概念可追溯至1980年,直至2012年Dixon团队才正式确立其命名体系[3]。该过程的分子特征表现为细胞内二价铁离子超载触发双重效应。一方面通过芬顿反应催化多不饱和脂肪酸(polyunsaturated fatty acid, PUFA)过度氧化,促进单不饱和脂肪酸(monounsaturated fatty acid, MUFA)合成,另一方面干扰线粒体氧化磷酸化过程,导致活性氧(reactive oxygen species, ROS)异常累积。当ROS突破抗氧化防御阈值时,丙二醛(malondialdehyde, MDA)、4-羟基壬烯醛(4-hydroxynonenal, 4-HNE)等脂质过氧化物级联生成,最终引发细胞膜系统崩解[5]。铁死亡相关分子机制图见图1(铁死亡的分子机制)。区别于凋亡与坏死的典型形态学特征,铁死亡呈现出独特的线粒体病理改变——体积皱缩、嵴结构消失伴随双层膜致密化,但细胞核保持完整且无染色质固缩,该过程不受经典凋亡、坏死调控因子的影响,其特异性生化标志包括铁积累、谷胱甘肽耗竭及NADPH氧化应激[6]。
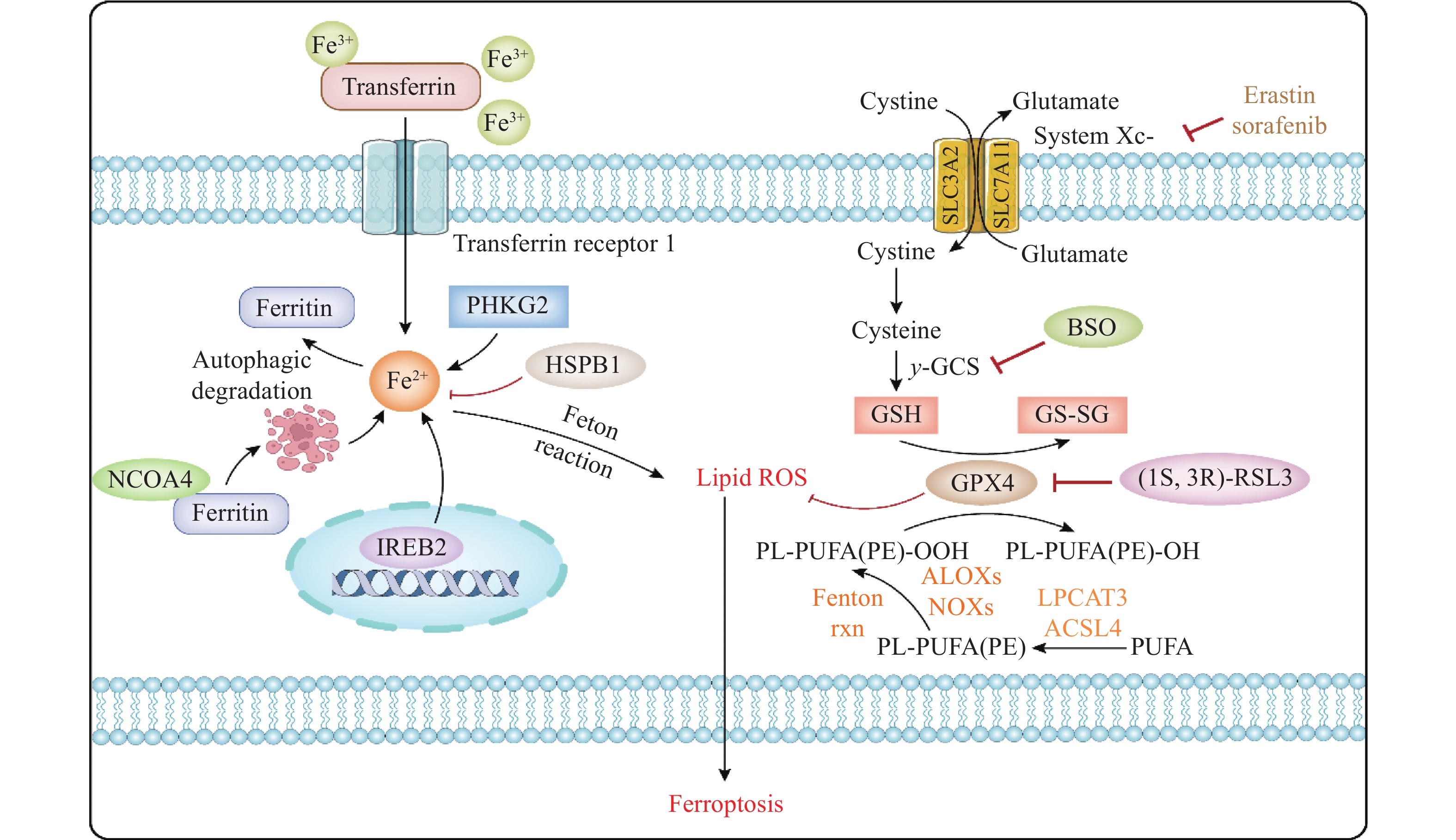
1.1 铁代谢失衡
铁死亡的核心驱动力源于非酶促性铁催化反应,其中GPX4失活引发的抗氧化防御崩溃尤为关键。在GPX4抑制状态下,磷脂氢过氧化物(phospholipid hydroperoxides, PLOOHs)的异常蓄积通过芬顿反应触发脂质过氧化级联放大,其与Fe2+/Fe3+反应产生的自由基成为铁死亡的分子指纹[7]。铁代谢稳态由铁调节蛋白1 (iron regulatory protein 1, IRP1)和铁调节蛋白2 (iron regulatory protein 2, IRP2)调控网络精密控制,涉及铁储存[8]、转运[9]及释放[10]的动态平衡。具体而言,转铁蛋白-转铁蛋白受体复合体内吞途径通过STEAP3金属还原酶介导的Fe3+还原及二价金属离子转运蛋白1转运实现铁摄取,而铁蛋白自噬机制则构成铁缓冲体系。铁输出障碍或储存蛋白表达下调均导致游离铁超载,进而通过芬顿反应产生高活性羟自由基(•OH),引发DNA断裂、蛋白质氧化及膜脂损伤[11]。
1.2 脂质代谢异常
PUFA的过氧化是铁死亡的关键标志,其中长链脂酰辅酶A合成酶4 (long-chain lipoyl coenzyme A synthetase 4, ACSL4)和脂氧合酶(lipoxygenase, LOX)是脂质过氧化的重要调控因子。铁死亡的执行核心在于失控的脂质过氧化级联反应,其病理进程呈现显著的膜脂代谢重编程特征[12]。关键分子事件始于PUFA的活化与修饰:ACSL4催化游离PUFA生成酰基辅酶A衍生物,经LPCAT3酯化后嵌入磷脂形成PL-PUFA,该过程通过Thr328位点磷酸化增强酶活性以促进促死亡脂质底物积累[13]。在生物活性铁的存在下,PL-PUFA可以通过酶促和非酶脂质过氧化反应转化为PLOOHs,若PLOOHs不能被有效中和,从而积累破坏质膜完整性,细胞就会死于铁死亡[14]。
1.3 胱氨酸/谷氨酸逆向转运体拮抗铁死亡的发生
GPX4是抑制铁死亡的核心酶,其活性依赖于谷胱甘肽(glutathione, GSH)。胱氨酸/谷氨酸逆向转运体(System Xc-)功能障碍会导致GSH耗竭,进而削弱GPX4的抗氧化能力[15]。System Xc-作为SLC7A11/SLC3A2异二聚体,通过1︰1交换胞外胱氨酸与胞内谷氨酸维持氧化还原稳态[16]。摄入的胱氨酸经还原生成半胱氨酸,随后与谷氨酸(glutamate, Glu)、甘氨酸(glycine, Gly)在GSH合酶催化下合成GSH,该三肽的活性巯基不仅直接清除ROS,更是GPX4酶催化功能的关键辅因子[17-18]。GPX4通过将GSH氧化为GSSG,将毒性PLOOHs还原为无害磷脂醇,从而阻断脂质过氧化级联[17, 19]。
2 铁死亡参与DR的病理机制
2.1 高糖诱导的铁代谢紊乱
大量临床和基础研究表明,DR患者存在显著的铁代谢失衡。Sulochana等[20]的早期临床研究发现,增殖性DR患者玻璃体中的铁水平较健康人群升高2.5倍,提示铁超载与DR严重程度密切相关。
首先,高血糖会直接破坏血红蛋白结构,导致血红素分子解离并释放游离铁[21]。同时,高糖环境激活视网膜Müller细胞中的血管紧张素Ⅱ(angiotensin Ⅱ, Ang-Ⅱ)表达,其水平较正常状态升高达10倍[22]。Ang-Ⅱ通过上调铁代谢相关基因,显著增强视网膜细胞对铁的摄取能力[23]。这一过程与转铁蛋白受体介导的铁内流形成协同效应,共同加剧细胞内铁超载。
铁离子在视网膜内主要富集于视网膜色素上皮层(retinal pigment epithelium, RPE)与光感受器,使其对铁死亡具有特殊易感性。铁蓄积通过氧化应激-炎症-血管新生轴驱动DR进展。Blasiak等[24]指出,视网膜老化导致ROS生成增加,而铁超载通过芬顿反应进一步放大氧化损伤。光感受器中富含的二十二碳六烯酸等PUFA成为ROS攻击的主要靶点,引发脂质过氧化链式反应,同时抑制脑源性神经营养因子表达[25-26]。
动物实验为铁代谢紊乱的致病作用提供了直接证据。Chaudhary等[27]在链脲佐菌素诱导的糖尿病小鼠模型中发现视网膜铁异常沉积。进一步研究发现,遗传性铁过载模型鼠在糖尿病状态下表现出更严重的神经元死亡、血管损伤和血视网膜屏障(blood-retinal barrier, BRB)渗漏。视网膜微血管内皮细胞作为BRB的主要结构成分,其程序性死亡将直接破坏BRB完整性,导致血清铁异常渗入视网膜组织。机制研究表明,铁超载通过激活G蛋白偶联受体信号通路,诱导RPE细胞过度表达肾素,进而促进Ang-Ⅱ生成[27]。Ang-Ⅱ不仅通过激酶插入域受体激活血管内皮生长因子(vascular endothelial growth factor, VEGF)受体,增强血管通透性和新生血管形成[28],还可直接上调VEGF表达,加重视网膜缺血、渗出及黄斑水肿[29]。
2.2 脂质过氧化
视网膜作为人体PUFA最富集的部位,其单位组织耗氧量居各器官之首[30]。这种独特的生化特性使脂质过氧化成为DR的关键病理机制。脂质过氧化包含酶促与非酶促两种反应途径。在酶促途径中,LOX家族催化PUFA磷脂自由基的形成,该自由基经羟自由基作用后转化为多不饱和脂肪酸磷脂氢过氧化物。此类产物通过破坏细胞膜及线粒体膜完整性,显著增加膜通透性,从而触发铁死亡进程[31]。ACSL4作为铁死亡关键效应分子,催化PUFA合成脂酰辅酶A,进而经LPCAT3酯化为膜磷脂[13]。这些富含不饱和键的磷脂在铁离子与脂氧合酶作用下发生脂质过氧化,最终触发RPE细胞铁死亡[32]。
在糖尿病(diabetes mellitus, DM)病理状态下,代谢紊乱与慢性炎症导致细胞内铁稳态失衡,同时伴随铁死亡抵抗系统功能障碍。这种双重损伤使视网膜微血管内皮细胞(retinal microvascular endothelial cell, RMEC)对高糖环境尤为敏感[33]。光感受器细胞功能高度依赖脉络膜毛细血管经RMEC及RPE传递的氧与营养供应。RMEC损伤与RPE细胞功能障碍将引发光感受器代谢剥夺性死亡[34]。
尽管DR中铁死亡的具体调控机制尚未完全阐明,现有研究已证实ACSL4的异常激活,以及多重抗氧化系统的功能障碍,共同驱动光感受器铁死亡进程,进而揭示铁代谢紊乱、光感受器损伤与DR病理进展间的分子关联。
2.3 线粒体功能障碍
线粒体是ROS的主要生成源,其膜结构富含的PUFA更易遭受ROS攻击,形成特异性线粒体脂质过氧化物(lipid peroxide,LPO) [35],其积累可损伤线粒体DNA(mitochondrial DNA, mtDNA),导致其编码的电子传递链亚基功能障碍,同时线粒体铁库在病理状态下会释放氧化还原活性铁,进一步催化自由基级联反应[36]。尽管线粒体通过铁转运蛋白/铁蛋白系统调控铁稳态,并依赖mt-GPX4酶及线粒体谷胱甘肽池中和过氧化脂质,但在糖尿病环境中,视网膜毛细血管细胞呈现显著异常:胞质与线粒体ROS协同升高、LPO蓄积加剧、游离铁离子堆积,伴随线粒体膜渗漏及膜电位受损[37-38]。
在DR中,高糖诱导的线粒体功能障碍与铁死亡之间存在复杂的交互网络。线粒体电子传递链(electron transport chain, ETC)是ROS的主要来源,其中复合物I、Ⅱ和Ⅲ的电子泄漏可将氧分子(O2)还原为超氧阴离子(O2-),并进一步转化为H2O2和•OH[39]。过量ROS不仅直接损伤mtDNA,还可降低线粒体膜电位,导致ETC解偶联和ATP合成障碍[40-41]。光感受器作为高代谢神经元,其功能高度依赖线粒体能量供应。因此,线粒体质量控制机制,即通过线粒体自噬清除受损线粒体并通过线粒体生物发生补充健康线粒体的过程,对维持视网膜稳态至关重要[42-43]。
高糖环境通过破坏这一平衡机制驱动铁死亡。Chang等[44]的研究揭示了线粒体自噬与铁死亡的直接关联:在胰腺β细胞中,使用线粒体解偶联剂激活线粒体自噬后,GPX4表达下降,脂质过氧化标志物水平升高,表明过度线粒体自噬通过削弱抗氧化防御促进铁死亡。类似机制在RPE细胞中同样存在。Singh等[45]发现,高糖通过上调硫氧还蛋白相互作用蛋白诱导线粒体功能障碍,导致受损线粒体堆积并触发溶酶体依赖性铁释放。
2.4 炎症与血管通透性增加
铁死亡通过激活核因子κB (nuclear factor-κB, NF-κB)信号通路,在DR中形成“炎症-氧化损伤”恶性循环。研究表明,铁死亡诱导的脂质过氧化产物可直接激活Toll样受体4(toll-like receptor 4, TLR4),进而通过MyD88依赖性途径触发NF-κB核转位[46]。活化的NF-κB通过转录调控促进白细胞介素-6 (interleukin-6, IL-6)和白细胞介素-1β (interleukin-1β, IL-1β)的释放[47]。IL-6激活信号转导和转录激活因子3 (signal transducer and activator of transcription 3, STAT3)信号通路,导致闭锁小带蛋白-1(zonula occludens-1, ZO-1)和闭锁蛋白(occludin)的磷酸化水平增加,破坏内皮细胞间连接复合物的稳定性,使FITC-dextran通透性增加[48-49];同时IL-6与VEGF形成正反馈环,刺激血管内皮细胞分泌基质金属蛋白酶9 (matrix metalloproteinases 9,MMP-9),降解基底膜成分,加速玻璃体积血[50-51]。IL-1β则通过上调NOD样受体热蛋白结构域相关蛋白3 (NOD-, LRR- and pyrin domain-containing protein 3, NLRP3)炎症小体,诱导Caspase-1依赖的焦亡途径,进一步释放炎症因子并促进视网膜神经节细胞凋亡[52-53]。
3 铁死亡相关治疗策略
近年来,铁死亡的分子调控机制逐渐明晰,其诱导剂与抑制剂的开发为多种疾病治疗提供了新方向。铁死亡诱导剂主要靶向细胞内抗氧化系统与铁代谢通路,目前已知的核心诱导剂包括两类:一类是System Xc-抑制剂,如柳氮磺吡啶、Erastin和丁硫氨酸亚砜亚胺等。这类药物通过阻断胱氨酸摄取,导致GSH合成受阻,显著降低细胞内GSH水平,从而削弱GPX4的抗氧化能力,引发脂质过氧化蓄积。另一类为GPX4直接抑制剂,如Ras选择性致死分子3 (Ras-selective lethal 3,RSL3)、铁死亡诱导剂7 (death-promoting inhibitor 7, DPI7) 和铁死亡诱导剂10 (death-promoting inhibitor 10, DPI10)等,其通过共价结合GPX4的硒代半胱氨酸活性中心,不可逆抑制其还原LPO的功能。此外,非经典诱导通路逐渐受到关注,例如索拉非尼通过激活P62-Keap1-NRF2轴抑制抗氧化系统,同时下调铁死亡抑制蛋白1-辅酶Q10通路(ferroptosis suppressor protein 1-coenzyme Q10 pathway, FSP1-CoQ10),形成双重促铁死亡效应;而青蒿素衍生物则利用其内过氧化物结构螯合游离铁,触发芬顿反应并特异性损伤线粒体膜脂质。表1列举了常见的铁死亡诱导剂及其关键作用机制。
|
分类 |
药物/成分 |
作用机制 |
参考文献 |
|
靶向System Xc-
|
Erastin |
抑制System Xc-,阻断胱氨酸摄取,降低GSH水平,削弱GPX4活性 |
[54] |
|
|
柳氮磺吡啶(Sulfasalazine) |
抑制System Xc-功能,减少GSH合成,增强脂质过氧化能力 |
[55] |
|
|
索拉非尼(Sorafenib) |
抑制System Xc-,同时激活P62-Keap1-NRF2通路,双重抑制抗氧化系统 |
[56] |
|
|
咪唑酮(IKE) |
高效抑制System Xc-,特异性诱导癌细胞铁死亡 |
[57] |
|
靶向GPX4 |
RSL3 |
共价结合GPX4活性中心,不可逆抑制其酶活性,导致LPO积累 |
[58] |
|
|
ML162/ML210 |
直接抑制GPX4,诱导脂质过氧化依赖性细胞死亡 |
[59] |
|
|
FIN56 |
激活角鲨烯合酶,促进GPX4蛋白降解,同时消耗辅酶Q10,增强氧化损伤 |
[60] |
|
调节铁代谢 |
青蒿琥酯(Artesunate) |
诱导铁蛋白自噬,释放游离铁,促进芬顿反应及脂质过氧化 |
[61] |
|
|
FINO2 |
氧化Fe2+生成Fe3+和ROS,同时直接氧化PUFA |
[62] |
|
|
拉帕替尼(Lapatinib) |
上调转铁蛋白受体,增加细胞内铁摄取,促进铁依赖性氧化应激 |
[63] |
|
靶向脂质代谢 |
Troglitazone/Rosiglitazone |
抑制ACSL4活性,阻断PUFA与磷脂结合 |
[64] |
|
|
Zileuton |
抑制LOX,减少LPO生成,但特定条件下可能通过反馈机制加剧氧化损伤 |
[65] |
|
靶向线粒体 |
BAY87-2243 |
抑制线粒体呼吸链复合物I,增加线粒体ROS释放,触发脂质过氧化 |
[66] |
|
|
盐霉素(Salinomycin) |
诱导线粒体膜电位崩溃,释放细胞色素C,协同铁死亡与凋亡通路 |
[67] |
|
多靶点诱导剂 |
顺铂(Cisplatin) |
结合并消耗GSH,抑制GPX4活性;同时诱导DNA损伤,激活氧化应激信号通路 |
[68] |
|
|
姜黄素(Curcumin) |
抑制GPX4,激活Nrf2通路,在特定浓度下通过铁超载增强脂质过氧化 |
[69] |
在抑制策略方面,铁死亡抑制剂通过多途径阻断病理进程,表2列举了常见的铁死亡诱导剂及其作用机制。脂质过氧化中和剂如Ferrostatin-1(Fer-1)和维生素E直接干预氧化链式反应:Fer-1通过清除脂质过氧化自由基(LOO•)和螯合Fe2+发挥协同保护作用,而维生素E作为脂溶性抗氧化剂嵌入细胞膜,抑制PUFA的过氧化进程。铁代谢调节剂如去铁胺则通过降低游离铁水平减少芬顿反应驱动的ROS生成,临床前研究证实其可缓解索拉非尼诱导的肝细胞癌铁死亡,并在神经退行性疾病模型中保护神经元。抗氧化系统激活剂则通过增强内源性防御机制发挥作用,例如多巴胺和放线菌酮分别通过上调GPX4表达或激活System Xc-提升GSH合成能力,而硒化合物通过整合入GPX4提升其酶活性,例如硒强化蛹虫草与阿奇霉素联用可增强抗肿瘤疗效。
|
分类 |
药物/成分 |
作用机制 |
参考文献 |
|
铁螯合剂 |
去铁胺(Deferoxamine) |
减少游离铁蓄积,抑制芬顿反应,降低脂质过氧化水平 |
[70] |
|
|
锌-去铁胺(Zn-DFO) |
靶向清除RPE细胞中的铁,减轻光诱导损伤,保护RPE功能 |
[71] |
|
抗氧化剂 |
Fer-1/Liproxstatin-1 |
直接中和LPO,恢复细胞膜稳定性,阻断铁死亡级联反应 |
[72] |
|
|
黄芪甲苷-IV |
抑制miR-138-5p,上调Sirt1/Nrf2通路,增强GPX4、GCLM等抗氧化基因表达 |
[73] |
|
靶向信号通路 |
Gas6/MerTK/GPX4通路 |
Gas6通过激活MerTK抑制GPX4降解,减少ARPE-19细胞铁死亡,改善视网膜神经纤维层厚度 |
[74] |
|
|
TRIM46抑制剂 |
阻断TRIM46介导的IκBα泛素化,抑制NF-κB通路的促炎和促铁死亡作用,降低HRCECs通透性 |
[75] |
|
天然产物 |
葛根素(Puerarin) |
纠正视网膜铁超载,抑制TXNIP介导的线粒体氧化损伤,恢复线粒体膜电位 |
[76] |
|
|
白藜芦醇(Resveratrol) |
通过下调SLC1A5和ACSL4表达,减少PUFA-PE合成,减轻脂质过氧化 |
[77] |
|
|
槲皮素(Quercetin) |
抑制ACSL4活性,增强GPX4稳定性,协同阻断铁死亡和凋亡通路 |
[78] |
这些调控分子的转化潜力已在疾病模型中显现:诱导剂可针对铁死亡敏感的恶性肿瘤增强治疗效果,而抑制剂在神经退行性疾病、缺血再灌注损伤中展现出细胞保护作用。未来研究需进一步解析组织特异性调控网络,优化药物递送系统,以实现精准的时空动态干预,推动铁死亡靶向治疗从基础研究向临床转化迈进。
4 总结与展望
本研究基于铁死亡的分子调控机制及其在DR中的研究证据,系统分析该疾病与铁死亡之间的病理关联,重点揭示高糖诱导的铁代谢紊乱、脂质过氧化、线粒体功能障碍及炎症在该疾病发生发展中的协同作用,并列举了铁死亡的诱导剂和抑制剂,以期为进一步探索DR的发病机制与防治手段提供新思路。研究表明,铁蓄积通过氧化应激-炎症-血管新生轴驱动DR进展,铁超载诱导的氧化还原失衡及膜脂过氧化级联反应可能通过RPE细胞损伤、血管内皮屏障破坏等机制参与DR进展。基于此,已有多种铁死亡调节剂进入临床前研究阶段,通过靶向抑制脂质过氧化或铁蓄积展现出视网膜保护潜力。
未来需进一步研究DR中铁死亡通路的精细化调控机制。解析铁代谢动态平衡与氧化应激网络在疾病不同阶段的协同效应,阐明其驱动视网膜细胞铁死亡的分子级联规律。同时筛选高糖微环境特异性生物标志物,并建立其与疾病临床分期的定量关联模型,以提升病理进程监测的精准度,为DR的分层诊疗体系构建及靶向药物开发提供理论依据。
声明
本稿件在研究和论文撰写中未使用生成式人工智能,所有作者对内容的真实性、完整性和科学性负责。所有科学贡献和智力劳动均由所有作者共同完成。
利益冲突
所有作者均声明不存在利益冲突。
开放获取声明
本文适用于知识共享许可协议(Creative Commons),允许第三方用户按照署名(BY)-非商业性使用(NC)-禁止演绎(ND)(CC BY-NC-ND)的方式共享,即允许第三方对本刊发表的文章进行复制、发行、展览、表演、放映、广播或通过信息网络向公众传播,但在这些过程中必须保留作者署名、仅限于非商业性目的、不得进行演绎创作。