


HIGHLIGHTS
•Patients with von Hippel-Lindau (VHL) disease may have multi-system involvement including ocular lesions that decrease their quality of life. Severe visual impairment can occur due to retinal hemangioblastomas occuring at the optic nerve head and sequelae from large peripheral lesions, which may be difficult to treat with laser photocoagulation or cryotherapy.
•Numerous basic science experiments and several clinical trials have shown that hypoxia-inducible factor-2α (HIF-2α)inhibition causes regression of hemangioblastomas. Belzutifan is an oral HIF-2α inhibitor and based upon a phase 2 trial,belzutifan is now U.S. Food and Drug Administration (FDA) approved for the treatment of VHL-associated renal cell carcinoma, central nervous system hemangioblastoma, pancreatic neuroendocrine tumor, and endolymphatic sac tumor.
•Future directions include further characterization of the role of belzutifan in treating patients with ocular VHL lesions, including duration and dosage of therapy.
INTRODUCTION
Von Hippel-Lindau (VHL) disease is a rare genetic disease characterized by a propensity to develop benign and malignant tumors in multiple organs. It is inherited as an autosomal dominant trait with variable penetrance and its incidence is estimated around 1:36,000 livebirths. However, this dominant trait is also considered highly penetrant by age 60. This disease is caused by inactivating variants of the tumor suppressor VHL gene that plays a pivotal role in the hypoxia-inducible factor (HIF) pathway responsible for regulating cellular responses to hypoxia. The VHL gene is located on chromosome 3p25-26[1] and encodes the VHL protein that is involved in the ubiquitination and degradation of HIF. Disruption of this pathway leads to the accumulationof HIF, which induces angiogenesis and oncogenesis. Apositive family history can be present in 80% of cases, whereas de novo mutations occur in the remaining 20% of cases. To this day, more than 1,500 variants have been identified as causing VHL.[2]
VHL disease can manifest with a broad spectrum of highly vascularized tumors and cysts. VHL is diagnosed when either there is a positive family history for VHL and the presence of one or more VHL manifestations or when there are two or more VHL manifestations in patients without family history in the absence of genetic testing. Once a genetic variant is identified, it is crucial to test other family members who might carry the mutation.VHL-defining manifestations include hemangioblastoma in the retina or the central nervous system (CNS), renalcell carcinoma (RCC), pheochromocytoma, pancreatic neuroendocrine tumor (PNET), and endolymphatic sactumor (ELST). VHL patients can present with otherless specific clinical manifestations that are not part ofthe diagnostic criteria; these include simple pancreatic cysts, kidney cysts, and papillary cystadenoma of theepididymis or of the broad uterine ligament.[3]
VHL disease can manifest with a broad spectrum of highly vascularized tumors and cysts. VHL is diagnosed when either there is a positive family history for VHL and the presence of one or more VHL manifestations or when there are two or more VHL manifestations in patients without family history in the absence of genetic testing. Once a genetic variant is identified, it is crucial to test other family members who might carry the mutation.VHL-defining manifestations include hemangioblastoma in the retina or the central nervous system (CNS), renalcell carcinoma (RCC), pheochromocytoma, pancreatic neuroendocrine tumor (PNET), and endolymphatic sactumor (ELST). VHL patients can present with otherless specific clinical manifestations that are not part ofthe diagnostic criteria; these include simple pancreatic cysts, kidney cysts, and papillary cystadenoma of theepididymis or of the broad uterine ligament.[3]
Ophthalmologists play a key role in the management of patients with VHL disease, as ocular manifestations of the disease, namely retinal hemangioblastomas (RHs), are among the most common lesions, present in 49%-62% of persons affected with VHL.[4] We present an account of the basic and clinical research, including preclinical and clinical trials conducted at the National Eye Institute (NEI), National Institutes of Health (Bethesda, MD, USA), that have led to our current treatment paradigm for VHL-related RH.
BASIC SCIENCE EVIDENCE
Pathologic findings
RHs frequently are the initial and among the most common signs of VHL disease.[4-5] These tumors often present as a single vascular growth in any location in the retina but frequently in the superotemporal area of the peripheral retina, with the mean age of presentation of RH in the mid-twenties.[6-7] (Figure 1)
In a study conducted at the NEI, 205 out of 406 (50.5%) individuals with VHL developed eye-related conditions. Among the 205 patients, optic nerve hemangioblastomas were present in 48 (23.4%) patients, representing 11.1% of the entire group of VHL patients.[8] Optic nerve hemangioblastomas and juxtapapillary hemangioblastomas are rare conditions that progress slowly and typically have a poor prognosis (Figure 2). McCabe and colleagues observed that patients with juxtapapillary capillary hemangioblastoma and VHL often present at a younger age, tend to have tumors withan endophytic growth pattern, and frequently exhibit bilateral, multiple lesions.[9]
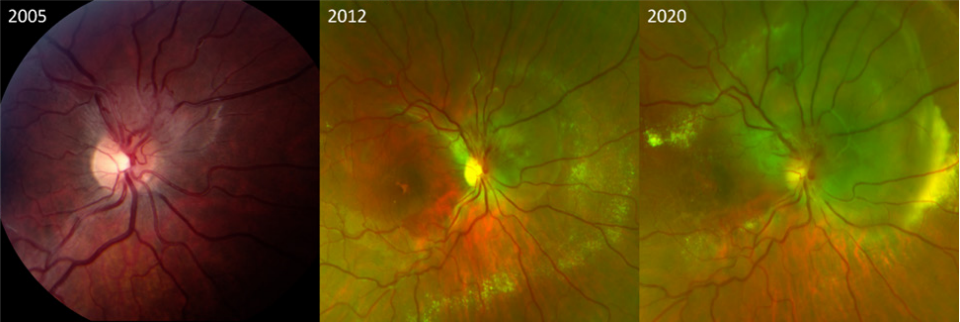
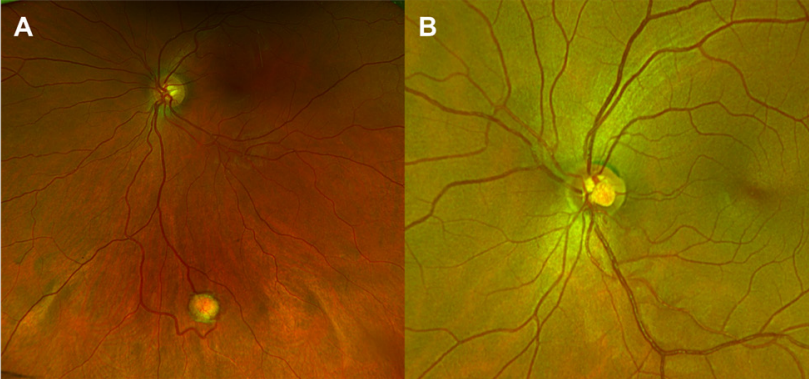
Figure 1 Color fundus photographs of retinal hemangioblastomas
The left image (figure A) displays a peripheral retinal hemangioblastoma in the inferior region of the retina in a VHL patient, accompanied by tortuous vessels supplying the tumor. The right image (figure B) depicts a juxtapapillary hemangioblastoma in another patient with VHL.In a study conducted at the NEI, 205 out of 406 (50.5%) individuals with VHL developed eye-related conditions. Among the 205 patients, optic nerve hemangioblastomas were present in 48 (23.4%) patients, representing 11.1% of the entire group of VHL patients.[8] Optic nerve hemangioblastomas and juxtapapillary hemangioblastomas are rare conditions that progress slowly and typically have a poor prognosis (Figure 2). McCabe and colleagues observed that patients with juxtapapillary capillary hemangioblastoma and VHL often present at a younger age, tend to have tumors withan endophytic growth pattern, and frequently exhibit bilateral, multiple lesions.[9]
Figure 2 Progression of right optic nerve hemangioblastoma over 15 years without treatment
Serial fundus photographs from 2005, 2012 and 2020 show the gradual progression of a right optic nerve hemangioblastoma with increasing exudation and macular involvement. Visual acuity declined from 20/20 in 2005 to 20/40 in 2012, and to 20/500 in 2020. The progression was exacerbated by pregnancy.Retinal and optic nerve hemangioblastomas exhibit similar histopathological characteristics to CNS hemangioblastomas. They consist of numerous abnormal capillary-like fenestrated channels encased by vacuolated foamy cells. These cells contain large lipid inclusions within their cytoplasm that are uniform with medium electron density. The tumors typically lack endothelial cells, but some may include reactive glial cells.[10-11]
In a study of histopathology of eight eyes from seven patients with a family history and clinical diagnosis of VHL disease, Chan et al.[10] examined the local genetic expression of the various aspects of the tumors. All specimens showed classic RHs associated with VHL,featuring numerous small capillary-like vascular channels interspersed with vacuolated stromal cells. RHs, like CNS hemangioblastomas, are composed of intermingled "stromal" and vascular cells. Tissue analysis suggests that "stromal" cells are genetically altered, retaining VHL gene deletions, while vascular cells maintain heterozygosity. In this study, samples rich in "stromal" cells consistently showed VHL deletions, whereas vascular samples did not. This indicates that the VHL gene deletion is specific to "stromal" cells, implicating them as the neoplastic component in RHs.
In a study of histopathology of eight eyes from seven patients with a family history and clinical diagnosis of VHL disease, Chan et al.[10] examined the local genetic expression of the various aspects of the tumors. All specimens showed classic RHs associated with VHL,featuring numerous small capillary-like vascular channels interspersed with vacuolated stromal cells. RHs, like CNS hemangioblastomas, are composed of intermingled "stromal" and vascular cells. Tissue analysis suggests that "stromal" cells are genetically altered, retaining VHL gene deletions, while vascular cells maintain heterozygosity. In this study, samples rich in "stromal" cells consistently showed VHL deletions, whereas vascular samples did not. This indicates that the VHL gene deletion is specific to "stromal" cells, implicating them as the neoplastic component in RHs.
Chan et al[12] analyzed specimens from four patients with typical VHL-associated ocular hemangioblastomas-three with intracranial optic nerve hemangioblastomas and one with juxtapapillary hemangioblastomas. Histologically, optic nerve hemangioblastomas resembled CNS hemangioblastomas more than retinal ones, featuring small vascular components with hemorrhages and tightly packed channels, which complicates the identification of vacuolated stromal cells. This finding may contribute to the aggressive nature of VHL lesions in the optic nerve. VHL disease, caused by a VHL gene mutation, shows loss of heterozygosity (LOH) in optic nerve hemangioblastomas, similar to other VHL tumors. Chan and colleagues found LOH in three of four cases, with overexpression of HIF-1 and ubiquitin secondary to loss of VHL protein function. LOH was present in stromal cells but not in endothelial cells, consistent with findings in VHL-related RHs. The study highlights that VHL lesions in the retina and optic nerve are primarily stromal cell tumors, with secondary vascularization due to increased vascular endothelial growth factor (VEGF) production.
Loss of heterozygosity of the VHL gene
VHL disease is a highly penetrant autosomal dominant condition. The disorder is caused by germline mutations in the VHL gene.[13] Typically, affected individuals inherit one mutant VHL allele from an affected parent and one normal allele from an unaffected parent. In other words, they are VHL heterozygous, carrying one mutated allele and one normal allele (VHL+/−). According to Knudson's two-hit hypothesis, the disease manifests when the second, normal, allele is lost or inactivated.[14-16] The "second hit" can occur through various mechanisms, such as deletion, point mutation, frameshift mutation, or promoter hypermethylation.[17-18] In about 20% of cases, the disease is sporadic, with somatic loss-of-function VHL mutations and no family history of the condition.[19] Tumor development in VHL disease is associated with the inactivation or loss of the remaining wild-type VHL allele in susceptible cells, leading to loss of the VHL protein (VHL−/−). This inactivation, whether due to mutation or hypermethylation, is commonly observed in non-hereditary clear RCCs and hemangioblastomas. In these cases, VHL gene alterations are somatic rather than inherited.[20]
Defect is the loss of the VHL protein
The VHL gene is relatively small, consisting of 854 nucleotides spread across three exons that code for the VHL protein (pVHL).[1] This protein is expressed in many cell types throughout the body and is involved in several regulatory pathways, including transcription, apoptosis, and extracellular matrix formation. Its primary role is facilitating the body's adaptation to hypoxic conditions.[21-22]
In normal oxygen conditions (normoxia), pVHL forms a complex with elongin B, elongin C, cullin-2 (Cul2), and Rbx1. This complex is essential for the ubiquitin-mediated degradation of HIFs like HIF-1α,HIF-2α, and HIF-3α.[23-25] (Figure 3)
In normal oxygen conditions (normoxia), pVHL forms a complex with elongin B, elongin C, cullin-2 (Cul2), and Rbx1. This complex is essential for the ubiquitin-mediated degradation of HIFs like HIF-1α,HIF-2α, and HIF-3α.[23-25] (Figure 3)
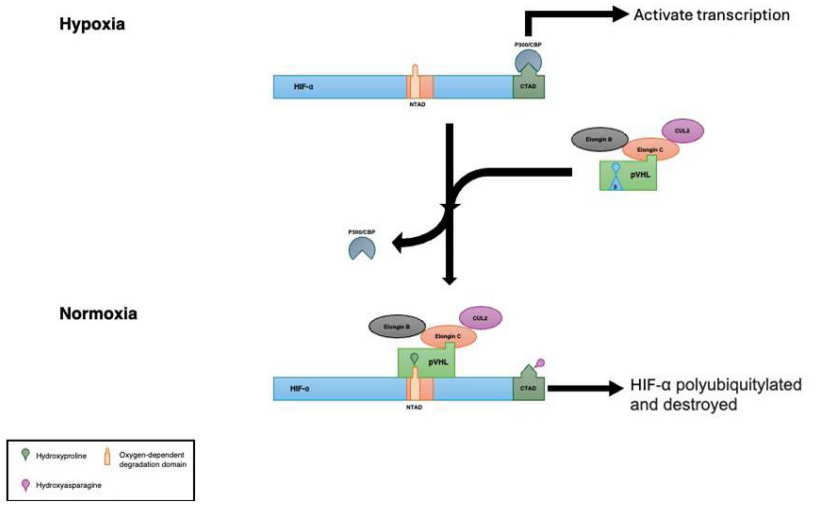
Figure 3 This image illustrates how pVHL regulates gene expression under normoxic and hypoxic conditions
pVHL controls hypoxia-inducible genes by binding to Hypoxia-Inducible Factor (HIF) and tagging it for degradation.Under low oxygen conditions (hypoxia), HIFα subunits become stabilized and accumulate in the cell, then move to the nucleus to bind with HIFβ. Thisinteraction triggers the activation of genes responsive to hypoxia, such as VEGF, platelet-derived growth factor-β (PDGF-β), erythropoietin (EPO), transforming growth factor-α (TGF-α), epidermal growth factor receptor,glucose transporter-1, and numerous others regulated by the HIF pathway. [21]
CXCR4, a HIF-induced protein, is usually suppressed by pVHL in normal oxygen levels. In contrast, during hypoxia, CXCR4 is activated by HIF. This activation suggests that CXCR4 may influence the attraction, migration, and development of embryonic VHL cells in the eye.[26]
Mutations in VHL lead to a state of "pseudohypoxia", characterized by elevated HIF levels and the subsequent activation of HIF-dependent genes. This activation promotes angiogenesis, enhances cellular proliferation, and shifts metabolism toward glycolysis. Additionally, non-HIF-dependent functions of pVHL are also involved in the disease's pathophysiology.[27-28]
William Kaelin’s contribution to HIF activity leading to the Nobel Prize (2019)
Research into the oxygen sensor regulating HIF activity advanced into the late 1990s. It was found that, under hypoxia, HIF-1α protein levels increase significantly without a corresponding rise in mRNA, indicating regulation at the protein level.[29-30] It was later established that, in oxygen-rich conditions, HIF-1α is degraded via an oxygen-dependent degradation domain (ODD) through the proteasome pathway.[31-32] Meanwhile, Kaelin was studying VHL disease, which results from mutations in the VHL tumor suppressor gene.[33] He suggested that VHL could influence the stabilization of hypoxia-inducible mRNAs, such as VEGF, in cancer cells. However, the exact mechanism by which VHL regulates its interaction with HIF-1α in response to oxygen levels remained unclear.[34-35] The key discovery was made by the Ratcliffe lab, which revealed that HIF-1α is marked for proteasomal degradation by an E3 ubiquitin ligase complex containing the pVHL when oxygen and iron are present. This crucial observation was subsequently validated by the Kaelin lab and other research teams.[36-38]
In 2001, Kaelin and Ratcliffe's teams made a critical breakthrough by revealing that the prolyl hydroxylation of HIF-1α, regulated by oxygen, enables its interaction with the pVHL. Ratcliffe's group identified three types of prolyl hydroxylase domains (PHDs) and showed that these enzymes need molecular oxygen to function effectively in modifying HIF-1α.[39-40]
We now understand that when intracellular oxygen levels fall, HIF-1α undergoes prolyl hydroxylation by PHDs, leading to its degradation by increasing its affinity for pVHL. Under hypoxia, this hydroxylation is suppressed, allowing HIF-1α to enter the nucleus and form a complex with HIF-1β and co-activators such as p300. This complex binds to hypoxia response elements in gene promoters, influencing various processes including angiogenesis, erythropoiesis, and glucose metabolism. The HIF-mediated response is now recognized as a key factor in disease outcomes, including cancer and inflammatory diseases.[41-44]
NATURAL HISTORY STUDY
A longitudinal study conducted at the NEI examined the progression of ocular VHL disease in a cohort of 249 participants over an average follow-up period of 8.2 years.[45] This study aimed to understand the natural history of ocular VHL disease and identify key risk factors associated with its progression. The participants, who had undergone multiple ophthalmic evaluations, were drawn from an initial group of 898 individuals with clinically defined systemic VHL disease.
At baseline, 62% of the participants exhibited clinical signs of ocular VHL disease in at least one eye,with 33% having bilateral involvement. Genetic analysis revealed that 96% of the participants had a documented germline mutation in the VHL gene. Missense mutations were the most common (48%), followed by protein- truncating mutations (42%), complete deletions (5%),and splice or amino acid deletions (5%).
At baseline, 62% of the participants exhibited clinical signs of ocular VHL disease in at least one eye,with 33% having bilateral involvement. Genetic analysis revealed that 96% of the participants had a documented germline mutation in the VHL gene. Missense mutations were the most common (48%), followed by protein- truncating mutations (42%), complete deletions (5%),and splice or amino acid deletions (5%).
Among the 95 participants who were free of ocularVHL disease at the baseline visit, 73% remained disease-free by the final follow-up, while 27% developed ocularmanifestations (n=26, with 18 experiencing unilateraldisease and 8 bilateral involvement).
For the natural history of RHs, 88% of susceptible eyes did not develop new RHs or progress to severe anatomical changes. Furthermore, 70% of eyes-maintained stability in the number of peripheral RHs, and 79% did not show increased RH involvement. However, 7% of eyes progressed to an end-stage phenotype characterized by severe anatomic involvement, such asphthisis or enucleation.
Visual acuity outcomes differed notably betweeneyes with pre-existing ocular VHL disease and those without. Eyes without pre-existing RH demonstrated a smaller mean change in best corrected visual acuity (BCVA) compared to those with disease (P = 0.0003).Eyes that developed new peripheral RHs experienced a slight decrease in BCVA (−2.3±1.2 letters), similar to eyes that remained disease-free (P = 0.86). In contrast, eyes with new juxtapapillary tumors experienced a more significant reduction in visual acuity (−12.7±5.2 letters)during mean follow-up of 8.24 years.
Key risk factors for anatomic progression and visual acuity decline included younger age at baseline, early onset of ocular VHL disease, and bilateral ocular involvement. Although the genotype of the germline VHL mutation was not significantly correlated with disease progression in traditional analyses, Kaplan- Meier time-to-event analyses indicated a lower risk of RH development in individuals with complete deletion mutations, compared to those with missense or protein- truncating mutations (P = 0.01).
This study highlights the complexity of ocular VHL disease progression and emphasizes the role of genetic and clinical factors in shaping individual patient outcomes. These findings underscore the importance of personalized monitoring and management strategies to optimize visual prognosis in patients with VHL disease.[45]
CLINICAL TRIALS CONDUCTED AT THE NATIONAL EYE INSTITUTE
Pegaptanib
The first interventional clinical trial at the NEI forocular VHL studied the use of intravitreal pegaptanib in eyes with juxtapapillary or large peripheral RHs in patients with VHL. Pegaptanib is a pegylated aptamer and acts as selective VEGF165 antagonist, suppressing pathological neovascularization and reducing vascular permeability.[46] Pegaptanib was approved by the U.S. Food and Drug Administration (FDA) in 2004 for neovascular age related macular degeneration (AMD),but was later replaced by more effective therapies.In VHL, VEGF is believed to be a key molecule in driving the formation and growth of RHs,[47] as well as the exudative changes due to increased vascular permeability.[48] This was an open label, non-randomized, prospective, pilot study that included five participants who received pegaptanib 3 mg every 6 weeks for at least 6 injections (30 weeks). The primary outcome was defined as a change of 15 letters (3 lines) in BCVA using Early Treatment Diabetic Retinopathy Study (ETDRS) charts by one year after initiating therapy. The secondary outcomes were macular thickness on optical coherence tomography (OCT) and leakage intensity and area on fluorescein angiography (FA). Three participants withdrew from the protocol due to adverse events,namely retinal detachment and worsening macular edema with an increase in hard exudates. In the two participants who completed the study, macular thickness as well as hard exudates were reduced. BCVA remained stable in one patient and improved in the other. Pegaptanib did not decrease the amount of fluorescein leakage, nor the size of the RH lesions. The authors concluded that the therapeutic response of pegaptanib was limited, showing potential benefits primarily in small lesions and when used in combination with other treatments.
Ranibizumab
Ranibizumab is an intravitreal anti-VEGF drug that received approval from the FDA for neovascular AMD,macular edema from retinal vein occlusion, and diabetic macular edema in 2006, 2010 and 2012, respectively.Ranibizumab is a recombinant humanized monoclonal antibody Fab fragment that binds and inactivates VEGF-A. At the NEI, an open label, non-randomized,prospective, pilot study evaluated five patients with VHL and RHs representative of severe ocular disease or disease resistant to standard therapy. The participants received monthly intravitreal injections of ranibizumab 0.5 mg for 6 months, with a final study visit at four weeks after the final study injection.[49] The primary outcome was again improvement of 15 letters (3 lines) in BCVA using ETDRS charts and secondary outcomes were change in lesion size, exudation on clinical exam and on FA, and change of macular thickness on OCT. All study eyes had decreased visual acuity associated with retinal exudation at baseline. BCVA improved in two studyeyes but decreased in three. The size of RHs increased in one study eye and remained stable in three. Decrease in tumor size was observed in only one study eye.Exudation increased in two study eyes, decreased in two, and remained stable in one. Again, this study showed that the benefits of intravitreal anti-VEGF were limited, with minimal effects on tumor size, occurring only in small VHL-related RHs.
Sunitinib
Sunitinib is a molecule that inhibits over 80 different receptor tyrosine kinases (RTKs), including VEGF receptors, and has been demonstrated to block tumor growth or to induce tumor regression in vitro and in vivo.Sunitinib was approved by the FDA for advanced renal cell carcinoma and unresectable or metastatic progressive well-differentiated pNET, and as second line agent for gastrointestinal stromal tumors (GIST). Systemic sunitinib yielded a significant response in non-VHL RCC.[50]
A patient was prescribed oral sunitinib for metastatic pancreatic neuroendocrine and kidney tumors and was observed at the NEI Clinical Center for regular follow- ups, ranging for 4 to 6 months for a period of 9 years. An open-label non-controlled prospective pilot study that was conducted at the NEI[51] was halted due to severe systemic side effects that included hepatotoxicity,left ventricular dysfunction, hypertension, hemorrhagic events, and thyroid dysfunction. Among the three patients in this case series, treatment with sunitinib did not improve visual acuity or reduce the size of RH.Improvements in RH associated retinal edema were observed in two participants in the study.
The investigators concluded that systemic sunitinib might play a role in managing the exudation in advanced RHs, when ablative treatments have failed, but that severe systemic adverse effects limit the use of this medication.[46]
A patient was prescribed oral sunitinib for metastatic pancreatic neuroendocrine and kidney tumors and was observed at the NEI Clinical Center for regular follow- ups, ranging for 4 to 6 months for a period of 9 years. An open-label non-controlled prospective pilot study that was conducted at the NEI[51] was halted due to severe systemic side effects that included hepatotoxicity,left ventricular dysfunction, hypertension, hemorrhagic events, and thyroid dysfunction. Among the three patients in this case series, treatment with sunitinib did not improve visual acuity or reduce the size of RH.Improvements in RH associated retinal edema were observed in two participants in the study.
The investigators concluded that systemic sunitinib might play a role in managing the exudation in advanced RHs, when ablative treatments have failed, but that severe systemic adverse effects limit the use of this medication.[46]
BELZUTIFAN
The previous trials conducted at the NEI targeted mostly VEGF and, perhaps unsurprisingly, these were not highly effective, as basic science has demonstrated the need to control HIF, given that there are many factors regulated by HIF. Belzutifan was developed as a HIF- 2α inhibitor and, for the first time, the mechanism of disease in VHL may be fully addressed by this therapy.In a phase 2, open-label, single-arm trial of belzutifan for RCC associated with VHL disease, 61 patients were enrolled at 11 centers between May 2018 and March 2019.[52] Eligible participants were 18 years or older, with VHL disease confirmed by a germline VHL alteration and at least one measurable RCC tumor. For patients with non-RCC neoplasms, baseline assessments included radiologic imaging and ophthalmic evaluations.
Participants received 120 mg of oral belzutifan once daily. The median age was 41 years (range: 19 to 66).After a median follow-up of 21.8 months (range: 20.2 to 30.1), the median time to response in RCC was 8.2 months (range: 2.7 to 19.1). An objective response was observed in 49% of patients with RCC (95% confidence interval: 36 to 62). Additionally, 77% of patients with pancreatic lesions (47 out of 61) and 30% of those with central nervous system hemangioblastomas (15 out of 50) showed improvement. Notably, all 16 eyes evaluated in 12 participants with RH showed improvement.
Participants received 120 mg of oral belzutifan once daily. The median age was 41 years (range: 19 to 66).After a median follow-up of 21.8 months (range: 20.2 to 30.1), the median time to response in RCC was 8.2 months (range: 2.7 to 19.1). An objective response was observed in 49% of patients with RCC (95% confidence interval: 36 to 62). Additionally, 77% of patients with pancreatic lesions (47 out of 61) and 30% of those with central nervous system hemangioblastomas (15 out of 50) showed improvement. Notably, all 16 eyes evaluated in 12 participants with RH showed improvement.
The most common adverse events were anemia,reported in 90% of patients, and fatigue, reported in 66%. Seven participants discontinued treatment: four withdrew voluntarily, one stopped due to a treatment-related adverse event (grade 1 dizziness), one due to disease progression as assessed by the investigator, and one patient died from acute fentanyl toxicity.[52]
In a subgroup analysis of this study conducted at the NEI,[53] eight participants had additional multimodal eye assessments. Within this subgroup, 10 out of 24 RH in 8 eyes of 6 participants were found to be 500 µm or more in greatest linear dimension at baseline and were analyzed in greater detail. All 10 hemangioblastomas showed a mean area reduction of 15% or more by month 12, which further increased to 30% or more by month 24.[52]
The effects of belzutifan on a patient who had an optic nerve hemangioblastoma followed for 12 years at the NEI before receiving the drug demonstrated marked regression of the tumor with almost complete regression of the abnormal vasculature of the optic nerve tumor (Figure 4). Such a response for the treatment of optic nerve hemangioblastomas has never been seen in other treatments, including the use of photodynamic therapy, laser, or radiation. Belzutifan is the first drug to result in such regression of these persistent and usually recalcitrant tumors.
CONCLUSION
Correction notice
None
Acknowledgement
None
Author Contributions
(Ⅰ) Conception and design: Emily Chew, Tiarnan Keenan, Alisa T. Thavikulwat
(Ⅱ) Administrative support: Emily Chew
(Ⅲ) Provision of study materials or patients: N/A as this was a review
(Ⅳ) Collection and assembly of data: All authors
(Ⅴ) Data analysis and interpretation: All authors
(Ⅵ) Manuscript writing: All authors
(Ⅶ) Final approval of manuscript: All authors
(Ⅱ) Administrative support: Emily Chew
(Ⅲ) Provision of study materials or patients: N/A as this was a review
(Ⅳ) Collection and assembly of data: All authors
(Ⅴ) Data analysis and interpretation: All authors
(Ⅵ) Manuscript writing: All authors
(Ⅶ) Final approval of manuscript: All authors
Funding
All authors are currently supported by the intramural research program of the National Eye Institute. Patient consent for publication
Conflict of Interests
None of the authors has any conflicts of interest to disclose. All authors have declared in the completed the ICMJE uniform disclosure form.
Patient consent for publication
Not applicable
Ethical Statement
IRB approval for these secondary analyses were obtained at the National Eye Institute.
Provenance and Peer Review
This article was a standard submission to our journal. The article has undergone peer review with our anonymous review system.
Data Sharing Statement
None
Open Access Statement
This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license).