


HIGHLIGHTS
• What is already known on this topic–Type 3 macular neovascularization (MNV3) is one type of neovascular age-related macular degeneration (AMD). Human Vhl mutations induce VHL disease, including retinal capillary hemangioblastoma (RCH). Pathologically, RCH and MNV3 are similar. In contrast to humans, VhlKO in mouse retina suppresses retinal angiogenesis, but Rb/p107/Vhl TKO can form an MNV3-like lesion.
• What this study adds–Similar to Rb1, p107 can also bind and suppress the transcription of many Hif target genes in the VhlKO retina.
• How this study might affect research, practice, or policy-This result enhances our understanding of the pathogenesis of MNV3 and RCH. The crosstalk between the cell cycle and hypoxia pathways may be the key to these diseases.
Type 3 MNV is also called retinal angiomatous proliferation (RAP).[3] It is an important subtype of neovascular AMD. Yannuzzi et al.[4] named this disease RAP to suggest an intraretinal origin and proposed a three-stage model of progression, including intraretinal neovascularization (IRN, stage I), subretinal neovascularization (SRN, stage II), and choroidal neovascularization (CNV, stage III). MNV3 accounts for 15%–20% of wet AMD patients in white populations[5]and 4.5%–11.1% among Asians.[6] Pathologically, MNV3 are retinal glomerular angiomatous lesions with encapsulation.[7] These features are similar to intraocular-injected vascular endothelial growth factor (VEGF)-induced retinal phenotypes in adult primates[8] and von Hippel-Lindau (VHL) gene mutation-related retinal capillary hemangioblastoma (RCH), which mainly have proliferating endothelial cells and VHL-deficient foamy (lipid-filled) stromal cells.[9] While anti-VEGF therapy has achieved favorable outcomes in stages I and II MNV3,[10-11] these therapies can develop geography atrophy and RPE rip. The long-term visual outcome of stage III lesions is still poor,[12] and recurrences are expected after the cessation of treatment. [13]
While clinical studies have provided important clues toward the potential pathogenesis of MNV3, proper experimental animal models are required to elucidate underlying mechanisms. Based on pathological studies, retinal hypoxia and inflammation are involved in developing MNV3. VHL tumor suppressor is a component of the E3 ubiquitin ligase complex that targets hypoxia-inducible factor α (Hif-α) for proteasomal degradation, which is the master regulator of the response to hypoxia.[14-15] Human VHL gene mutations cause RCH.[9] Pathologically, RCH is similar to MNV3, including glomerular IRN [7,16-17] and intraretinal lipid-filled cells beside IRN.[18]
Several retinal-specific Vhl knockout mice have been established to mimic RCH, using different Cre mouse lines and Vhl floxed mice.[19] While these Vhl knockout (KO) retinas have upregulated Hif expression, paradoxically, high Hif does not induce extensive angiogenesis as expected but instead inhibits retinal angiogenesis in the murine Vhl null retina.[20-21] The mechanism is unknown, but we found some clues in our previous report.[22] The retinoblastoma protein Rb1 regulates cell cycle progression and is mutated in many cancer types, including retinoblastoma. Rb1 and the related proteins p130 and p107 are members of the pocket protein family, which binds to E2f transcription factors.[23] We showed that Rb1 constrains the expression of some Hif target genes in the Vhl -/- retina. Deleting Rb1 induced extensive retinal neovascularization in the Vhl -/- retina. However, the Rb/Vhl double knockout (DKO)retina still can’t form subretinal MNV3-like lesions; unexpectedly, triple knockout (Rb/p107/VhlTKO) mice generate subretinal vascular growths resembling MNV3 and RCH.[22]
Rb1 and p107 are similar in structure and have functional redundancy and compensation.[24-25] In this study, we utilized genetics, RNA sequencing, chromatin immunoprecipitation (CHIP), and luciferase reporter assays to investigate the role of p107 in this MNV3-like mouse model. Strikingly, p107 deletion in the Vhl -/-retina triggered extensive retinal neovascularization. This phenotype was linked to direct binding and repression of Hif target genes in the retina.
To determine whether cell death had occurred, the frozen retinal sections were labeled by TdT-dUTP terminal nick-end labeling (TUNEL)with an apoptosis detection kit (In Situ Cell Death Detection Kit; Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions.
For whole-mount staining, eyeballs were enucleated and incubated for 30 minutes in 4% paraformaldehyde. The retinas were incubated at 4°C with FITC conjugated-IB4 (Sigma, L2895) and DAPI in PBS for 1-2 days. After brief washes with PBS, radial cuts were made to divide the retina into four quadrants to flatten the retina, and flat retinas were mounted with Mowiol.
Stained sections and slides were analyzed using a Zeiss Axio Imager Z2 fluorescence microscope and a Nikon C1si confocal microscope. Image J was used for cell counting. The positive cells of TUNEL, Ki67, γ-H2ax, and cell type markers were counted manually. Representative images were analyzed using the AngioTool (NCI) for vascular blood vessel analysis. In brief, at least three ×200 magnification images (320 μm×320 μm fields of view [FOV] per retina) per eye and three eyes from the same genotypes of different litter were counted.
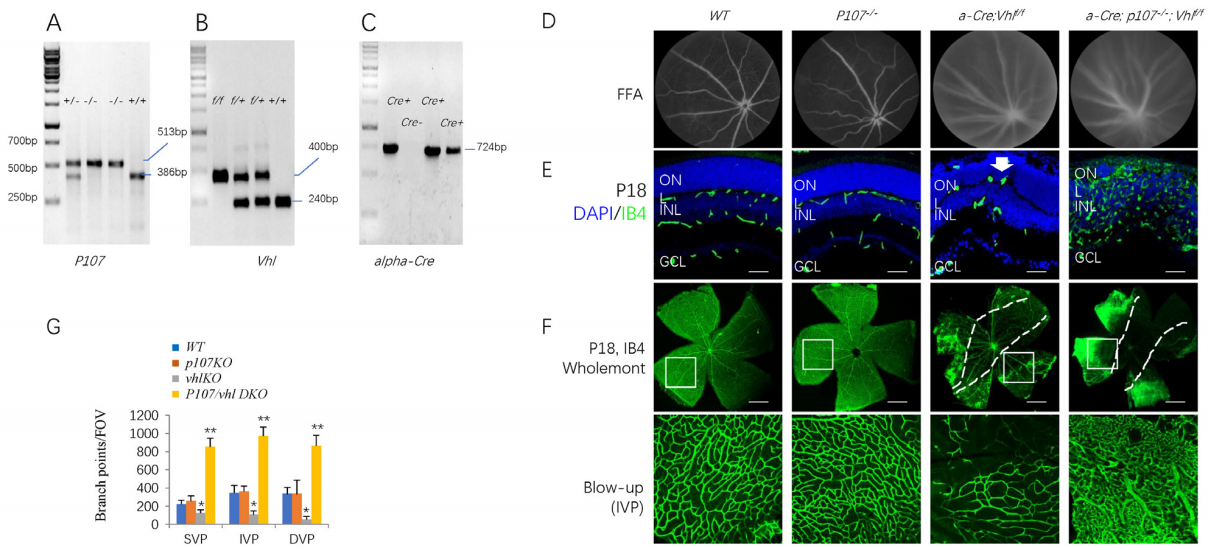
On the other hand, in the p107/VhlDKO retinas, there were many new blood vessels in the peripheral retina (Figure 1D-E). These vessels penetrated all retinal layers to form a dense capillary bed, and there was no normal SVP (superficial vascular plexus), IVP (intermediate vascular plexus), or DVP (deep vascular plexus) (Figure 1F). To compare the vascular density to other genotypes, we used vascular images from similar depths below the GCL (ganglion cell layer) to represent the IVP and DVP for the DKO retina. The branch points were 2-3 times higher than in WT, p107KO, and VhlKO retina (Figure 1G). Thus, deleting p107 unleashes the proangiogenic effects of Vhl loss in the retina.
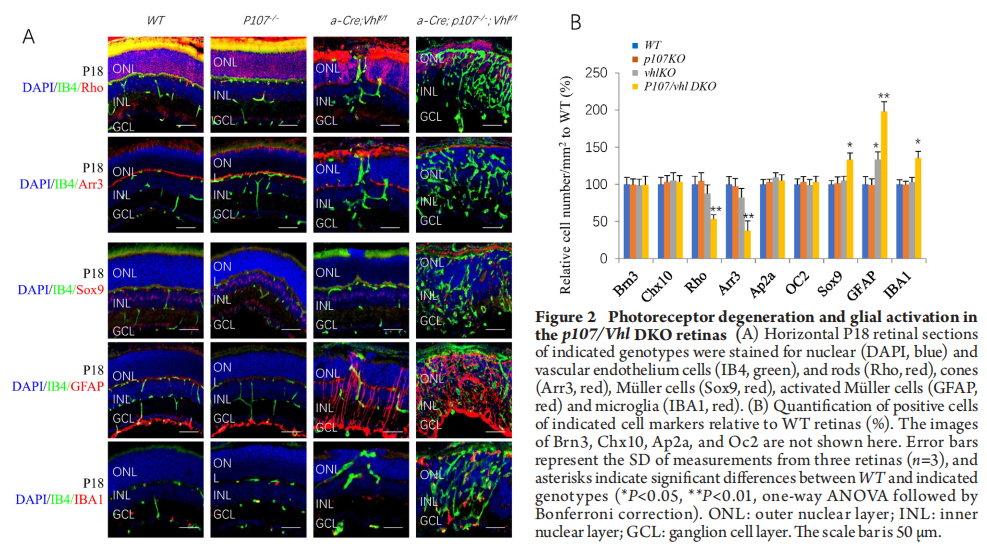 Cell loss or gain suggested an imbalance of cell proliferation and cell death. Ki67 staining (Figure 3A) found that VhlKO and p107/VhlDKO enhanced cell proliferation (Figure 3B). Still, most of these proliferative cells are IB4+ vascular endothelium cells (Figure 3C), and some are dividing Sox9+Müller cells in the DKO retinas (Figure 3C), but not IBA1+ microglia cells (Figure 3C). The p107/Vhl DKO also induced apoptosis (Figure 3A-B) and DNA damage (Figure 3A-B).
Cell loss or gain suggested an imbalance of cell proliferation and cell death. Ki67 staining (Figure 3A) found that VhlKO and p107/VhlDKO enhanced cell proliferation (Figure 3B). Still, most of these proliferative cells are IB4+ vascular endothelium cells (Figure 3C), and some are dividing Sox9+Müller cells in the DKO retinas (Figure 3C), but not IBA1+ microglia cells (Figure 3C). The p107/Vhl DKO also induced apoptosis (Figure 3A-B) and DNA damage (Figure 3A-B).
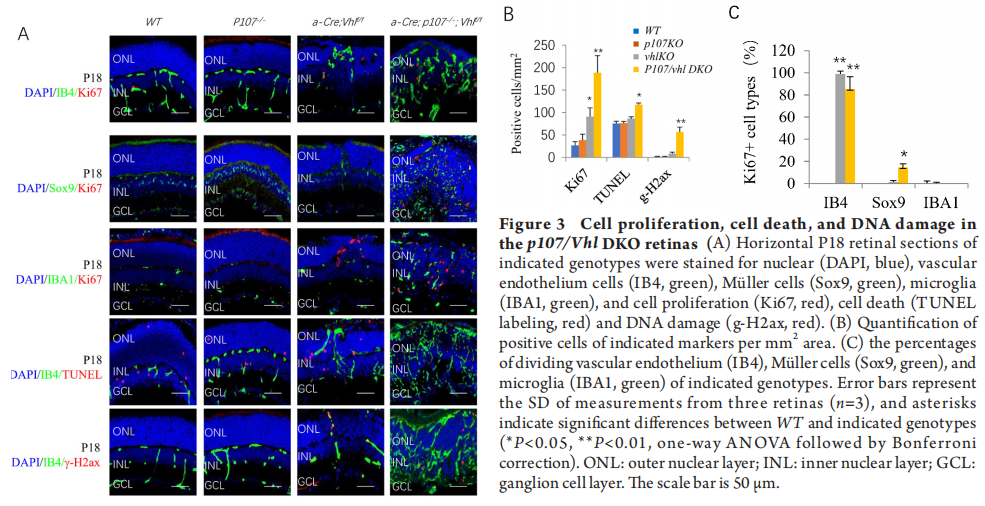
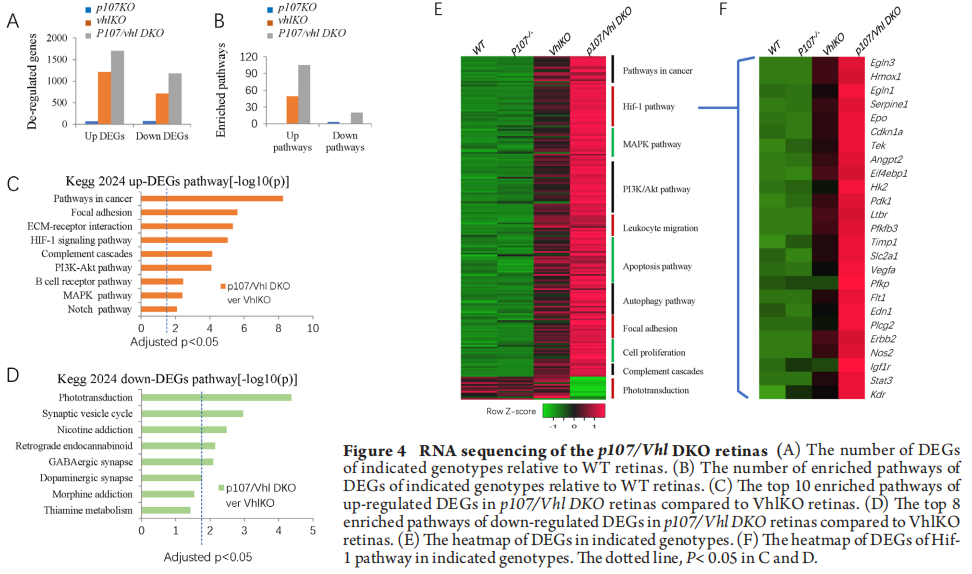
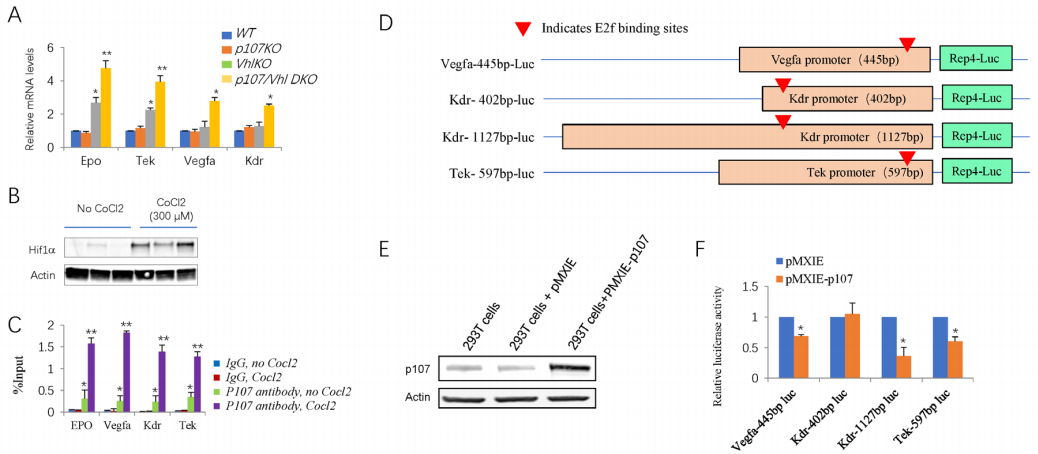
Figure 5 P107 protein can bind the promoter and suppress the transcription of several Hif target genes
We built four Rep4-luc-based luciferase reporter plasmids containing different sizes of promoter regions of Vegfa, Kdr, and Tek genes (Figure 5D).[22] P107 protein can be expressed in 293T cells transfected with the pMXIE-p107 construct (Figure 5E). Dual luciferase reporter assay using 293T cells indicated that p107 could partially repress the transcription activity of the 445bp-promoter of Vegfa gene (~34% reduced compared to empty vector), the 1127bp-promoter of Kdr gene (~64% reduced compared to the empty vector) and the 597bp-promoter of Tek gene (~40% reduced compared to the empty vector). P107 did not affect the shorter 402bp-promoter of Kdr (Figure 5F), suggesting a requirement for upstream regions. The result with the Vegfa 445bp-promoter-luc reporter is consistent with results showing that E2f1 can regulate a similar reporter. [31]
Together, the above genetic and molecular analyses reveal that p107 inhibits a subset of Hif1 target genes, providing a logical mechanism to explain why the Vhl -/-retina does not exhibit vessel overgrowth phenotypes, such as RAP or RCH.
While there is no evidence that Hif-1α can recruit Rb1 to the promoter of these Hif target genes,[22] our CHIP data supports the notion that Hif-1α can recruit p107 as CoCl2 treatment greatly enhanced the p107 binding to the promoter of Hif targets. It is well known that Rb1 preferentially interacts with E2f1-3, and p107 mainly binds to E2f4/5. P107 and E2f4/5 are both components of the DREAM complex(Dp, Rb-like, E2F4/5, and MuvB), which is a transcriptional repressor.[33]FoxM1, one component of the DREAM complex, is a downstream target of Hif-1α.[34] Thus, it is possible that Hif-1α recruit p107 through the DREAM complex.
Together, these data present a strong case that p107 directly inhibits Hif target expression in the retina and explains why vascular growth is constrained in the Vhl -/-murine tissue. Our work exposes previously unrecognized roles for the pocket protein family (Rb1 and p107) in constraining retinal angiogenesis, which will be essential to dissect the pathogenesis of MNV3 and RCH.
Besides our Rb/p107/VhlTKO mouse model, many studies use other Cre strains to knock out the Vhl gene in the mouse eye to study the mechanism of MNV3, RCH, or retinal neovascularization. The significant differences between our model and these models are in three aspects: different targeting cells, knockout strategies (single VhlKO or combined VhlKO with other gene knockouts), and potential cell-of-origin of these lesions. For instance, removing Vhl in murine hemangioblasts using Scl-CreER triggered many features of early-stage RCH, including dilated tortuous vessels, vascular leakage and foamy stromal cell clusters.[35] This mouse model is a single VhlKO; the knockout occurs in hemangioblasts, which can differentiate into vascular endothelium cells, and it suggests that embryonically arrested hemangioblasts are the cell-of-origin of RCH. In addition, this model can produce hemangioblastoma-like lesions in the mouse brainstem, like human VHL diseases.[35] This work is a milestone in the mouse RCH model field, but the lesions are relatively minor. Pathologically, they only present some “tumorlet” cells adjacent to the dilated vessels. It will be exciting to examine the expression of pocket proteins in these hemangioblasts and test if removing Rb1 or p107 can exaggerate their phenotypes.
Pde6g-CreERT2-mediated VhlKO in rod cells can cause retinal neovascularization, resembling the early stage of human MNV3.[35] However, PAS staining only identified very few subretinal new vessels. Thus, this model differs significantly from ours regarding the subretinal vascular phenotypes.[22] This mouse model is like other well-known models for early stage of MNV3, such as VhlKO in mouse RPE cells,[36] and models related to abnormal lipid metabolism (Vldlr-/- mice,[37] Cyp27a1-/- mice[38]), or abnormal cell migration pathway (endothelial cell-specific Srf knockout mice[39]), and RNV3 (JR5558) mice which have mutations in genes regulating retinal polarity and inflammation.[19,40]
In the mouse model of oxygen-induced retinopathy (OIR), LysmCre-mediated VhlKO in myeloid cells can promote the expression of VEGF and bFGF, enhance retinal vascular regeneration, and reduce pathological retinal neovascularization.[41] This model lacks MNV3/RCH-like lesions but reveals a complex interaction between myeloid cells and vascular endothelium cells under hypoxia. However, this work suggests that myeloid cells may not be the cells of origin of RCH, as myeloid VhlKO reduces pathological retinal neovascularization.
In summary, our MNV3/RCH-like mouse model is unique in three aspects. First, it is mainly based on the Pax6 α-Cre mediated VhlKO in retinal progenitors. Second, it knocked out the other two genes (Rb1 and p107) and the Vhl gene.[22] Similarly, Vhl loss alone does not induce kidney Vhl disease, but Rb1/p53/Vhl triple knockout induces a CCRCC (clear cell renal cell carcinoma) mouse model.[42] Based on these two works, it seems likely that while VHL mutation alone can induce VHL disease in humans (including RCH and CCRCC), it can’t cause VHL diseases in mice. Third, we show that the foamy cells in the MNV3/RCH-like lesions originate from Müller glia, which express high levels of Sox9 and Gal3. This is consistent with a report that Gal3 is a diagnostic marker for human brain hemangioblastoma,[43]which indicates that Gal3 is also a possible biomarker for diagnosing human RCH.[22]
(Ⅱ) Administrative support: Lirong Xiao
(Ⅲ) Provision of study materials or patients: Danian Chen
(Ⅳ) Collection and assembly of data: Ling Huang, Yiwen Hong,Wenyu Du, Wei Qiang, Wenyue Chen, Yujiao Wang, Ran Wei
(Ⅴ) Data analysis and interpretation: Ling Huang, Yiwen Hong, Wenyu Du, Danian Chen
(Ⅵ) Manuscript writing: All authors
(Ⅶ) Final approval of manuscript: All authors
INTRODUCTION
Age-related macular degeneration (AMD) is the leading cause of legal blindness in aged individuals, which can be divided into dry and wet AMD.[1] Dry AMD accounts for about 85%, manifests as degeneration of RPE (retinal pigment epithelium) cells and photoreceptors. Wet AMD accounts for about 15% of the cases and manifests as macular neovascularization (MNV). More than 80% of patients blinded by AMD are due to wet AMD.[2] MNV can be classified into type 1, type 2, and type 3 MNV.[3]Type 3 MNV is also called retinal angiomatous proliferation (RAP).[3] It is an important subtype of neovascular AMD. Yannuzzi et al.[4] named this disease RAP to suggest an intraretinal origin and proposed a three-stage model of progression, including intraretinal neovascularization (IRN, stage I), subretinal neovascularization (SRN, stage II), and choroidal neovascularization (CNV, stage III). MNV3 accounts for 15%–20% of wet AMD patients in white populations[5]and 4.5%–11.1% among Asians.[6] Pathologically, MNV3 are retinal glomerular angiomatous lesions with encapsulation.[7] These features are similar to intraocular-injected vascular endothelial growth factor (VEGF)-induced retinal phenotypes in adult primates[8] and von Hippel-Lindau (VHL) gene mutation-related retinal capillary hemangioblastoma (RCH), which mainly have proliferating endothelial cells and VHL-deficient foamy (lipid-filled) stromal cells.[9] While anti-VEGF therapy has achieved favorable outcomes in stages I and II MNV3,[10-11] these therapies can develop geography atrophy and RPE rip. The long-term visual outcome of stage III lesions is still poor,[12] and recurrences are expected after the cessation of treatment. [13]
While clinical studies have provided important clues toward the potential pathogenesis of MNV3, proper experimental animal models are required to elucidate underlying mechanisms. Based on pathological studies, retinal hypoxia and inflammation are involved in developing MNV3. VHL tumor suppressor is a component of the E3 ubiquitin ligase complex that targets hypoxia-inducible factor α (Hif-α) for proteasomal degradation, which is the master regulator of the response to hypoxia.[14-15] Human VHL gene mutations cause RCH.[9] Pathologically, RCH is similar to MNV3, including glomerular IRN [7,16-17] and intraretinal lipid-filled cells beside IRN.[18]
Several retinal-specific Vhl knockout mice have been established to mimic RCH, using different Cre mouse lines and Vhl floxed mice.[19] While these Vhl knockout (KO) retinas have upregulated Hif expression, paradoxically, high Hif does not induce extensive angiogenesis as expected but instead inhibits retinal angiogenesis in the murine Vhl null retina.[20-21] The mechanism is unknown, but we found some clues in our previous report.[22] The retinoblastoma protein Rb1 regulates cell cycle progression and is mutated in many cancer types, including retinoblastoma. Rb1 and the related proteins p130 and p107 are members of the pocket protein family, which binds to E2f transcription factors.[23] We showed that Rb1 constrains the expression of some Hif target genes in the Vhl -/- retina. Deleting Rb1 induced extensive retinal neovascularization in the Vhl -/- retina. However, the Rb/Vhl double knockout (DKO)retina still can’t form subretinal MNV3-like lesions; unexpectedly, triple knockout (Rb/p107/VhlTKO) mice generate subretinal vascular growths resembling MNV3 and RCH.[22]
Rb1 and p107 are similar in structure and have functional redundancy and compensation.[24-25] In this study, we utilized genetics, RNA sequencing, chromatin immunoprecipitation (CHIP), and luciferase reporter assays to investigate the role of p107 in this MNV3-like mouse model. Strikingly, p107 deletion in the Vhl -/-retina triggered extensive retinal neovascularization. This phenotype was linked to direct binding and repression of Hif target genes in the retina.
MATERIALS AND METHODS
Mouse strains and genotyping
α-Cre mice (P. Gruss), Rbl1 -/- (p107 -/-) mice (M. Rudnicki), and Vhl f/fmice (Jackson Laboratory, stock#004081) were maintained on a mixed background. All procedures were reviewed and approved by the Ethical Review Committee of Animal Research of West China Hospital, Sichuan University, Chengdu, China (AUP# 2020010A), and performed in compliance with the Association for Research in Vision and Ophthalmology (ARVO) statement for the use of animals in ophthalmic and visual research.The genotyping was performed as previously described.[22,24] The genotyping primers are listed in Table 1.Table 1 Genotyping primers
Histology, immunofluorescence and measurements
Eyeballs were fixed for 1 hour at 4°C in 4% paraformaldehyde, embedded in OCT (TissueTek 4583), frozen on dry ice, and cut into 12-14 µm sections on Superfrost slides. The following antibodies were used: Ap2a (Santa Cruz SC-8975), Arr3(Millipore AB15282), Brn3 (Santa Cruz SC-6062), γ-H2ax (Millipore, 05-636), GFAP (Abcam, Ab7260), IBA1 (Wako 019-19741), Ki67 (BD science Pharmingen 550609), Onecut2 (R&D system AF6294), Rhodopsin (Santa Cruz SC-57433), Sox9(Millipore AB5535). Vascular endothelial cells were labeled by FITC conjugated-IB4 (Sigma, L2895). Antigen retrieval was performed as described.[26] Primary antibodies or labeled cells were visualized using donkey anti-mouse, donkey anti-rabbit, and donkey anti-goat antibodies conjugated with Alexa-488, Alexa-568, or Alexa-647 (1:1000; Molecular Probes). Nuclei were counter-stained with DAPI (Sigma, D9542) and mounted with Mowiol.To determine whether cell death had occurred, the frozen retinal sections were labeled by TdT-dUTP terminal nick-end labeling (TUNEL)with an apoptosis detection kit (In Situ Cell Death Detection Kit; Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions.
For whole-mount staining, eyeballs were enucleated and incubated for 30 minutes in 4% paraformaldehyde. The retinas were incubated at 4°C with FITC conjugated-IB4 (Sigma, L2895) and DAPI in PBS for 1-2 days. After brief washes with PBS, radial cuts were made to divide the retina into four quadrants to flatten the retina, and flat retinas were mounted with Mowiol.
Stained sections and slides were analyzed using a Zeiss Axio Imager Z2 fluorescence microscope and a Nikon C1si confocal microscope. Image J was used for cell counting. The positive cells of TUNEL, Ki67, γ-H2ax, and cell type markers were counted manually. Representative images were analyzed using the AngioTool (NCI) for vascular blood vessel analysis. In brief, at least three ×200 magnification images (320 μm×320 μm fields of view [FOV] per retina) per eye and three eyes from the same genotypes of different litter were counted.
RNA sequencing
Total retinal RNAs were extracted using TRIzol (Invitrogen) and treated with RNase-free DNase I (New England Biolabs, Beverly, MA). The yield of total RNA was assessed using Nanodrop (Thermo). The cDNA libraries were prepared using the Illumina TruSeq RNA sample preparation kit, and the qualities were assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). For sequencing, the cDNA libraries were loaded on an Illumina HiSeq 2500 at Biomaker (Beijing, China). The raw sequence reads in fastq format were processed and analyzed. The expression fold change of each coding gene of mutated retinas (including Vhl -/-, p107-/-, p107/Vhl DKO) retina compared to WT retina was calculated using the following formula: Fold change=(RPKM+1)mutated/ (RPKM+1)wt. The genes, of which expression fold changes are greater than 1.54 or less than 0.65, were selected as the mutated-related differentially expression genes (DEG). The heatmap was generated by Heatmapper.[27] The function enrichment of DEG was performed using Enrichr,[28] the pathways with adjusted P < 0.05 were chosen to report.Quantitative real-time PCR
Total RNA was isolated from the peripheral retina (α-Cre expression area) using the Trizol Reagent (Invitrogen) followed by digestion with RNase-Free DNase (DNA-freeTM, Thermo Fisher Scientific) to remove DNA contamination. After quantification by a Nanodrop (NanoDrop Technologies, USA), first-strand cDNA was synthesized from 0.2-1µg of total RNA using the RT reagent Kit with gDNA Eraser (TaKaRa, China). PCR primers are listed in Table 2. Real-time quantitative PCR was performed using the qTOWER 2.2 PCR machine (Analytik Jena, Germany). Tests were run in duplicate on three separate biological samples with EvaGreen PCR Supermix (SsoFastTM, Bio-Rad laboratories, Singapore). Values obtained for test RNAs were normalized to β-actin mRNA levels.Table 2 RT-PCR primers
|
Genes |
Forward(5'-3') |
Reverse (3'-5') |
|---|---|---|
|
β-actin |
ACCACCACAGCTGAGAGGGA |
GCCATCTCCTGCTCGAAGTC |
|
Epo |
CTCCACTCCGAACACTCACA |
CCTCTCCCGTGTACAGCTTC |
|
Tek |
GAGGCCGAACATTCCAAGTA |
ATTGTCACATGGCCAAACAA |
|
Vegfa |
TCTTCCTGCCCACCATCTAC |
TGGTAACCCATGACCAGGAT |
|
Kdr |
AGTGGCTCTGTCCTCCAAGA |
TCTCACCCATCCTCAACACA |
Chromatin immunoprecipitation (ChIP)
Retinal explants obtained from postnatal day 8 wild-type C57/BL mice were cultured. Briefly, eyes were enucleated, retinas were carefully peeled away from RPE, and radial cuts were made to flatten the retina. The flattened retina was transferred to the membrane of a Millicell insert (Millipore, PICM03050) with the photoreceptors facing down. The insert was placed into the wells of a 6-well plate (Costar 3516, Corning), each contained 1,300 μL of retinal explant media, which was replaced every 2 days and was maintained in a 37 °C incubator with 5% CO2 for 3 days. The retinal explant basal medium was serum-free and made from Neurobasal A, DMEM/F12 medium, and N2/ B27 supplements (Life Technologies, USA). Cultured explants were exposed to 300 mol/L CoCl2 (Sigma, France), or kept as PBS-treated controls. The ChIP assay was performed using Magna ChIPTM A/G Chromatin Immunoprecipitation Kit (Millipore Temecula, USA) according to the manufacturer's instructions. Briefly, retinal explants treated with or without CoCl2 were cut into small pieces and then cross-linked for 20 min by adding formaldehyde to a final concentration of 1%. The cross-linking was stopped by adding 1/20 volume of 2.5 mol/L glycine and lysed by SDS lysis buffer. Then, cell lysate was sonicated and immunoprecipitated with antibodies specific to p107 (1:100, Santa Cruz, sc-318) or negative control IgG. After protein/DNA complexes were eluted, reverse cross-linked to free DNA, and purified, the specific DNA fragments were quantified using real-time PCR and normalized to input from the same sample. The primer sequences for the promoters analyzed are provided in Table 3.Table 3 CHIP primers
|
Promoter name |
Forward(5’-3’) |
Reverse (3’-5’) |
|
Epo promoter |
GGGGTGTGGCTCAGAAGTAG |
TGCAGTGCTGAGACTGGAAC |
|
Tek promoter Vegfa promoter |
GGCGATCTGGGTGTAAGAAA ATTCCTGGGAAAGGGAATTG |
GGAAGGCAATCAATTTGAGG AACTGGGCTCAGGAACACAC |
|
Kdr promoter |
GGCCAAAGCACCATAAAACA |
CAGTTGGGAGCATGAAGACA |
Luciferase reporter assays
The pREP4-Luc-based luciferase reporters were constructed as previously reported.[22] pMXIE and pMXIE-p107 vectors were from Dr. Rod Bremner.[23]293T cells (ATCC® CRL-11268™) were transfected with the pREP4-Luc vectors and the pMXIE and pMXIE-p107 vectors using Lipofectamine 2000 (Invitrogen). In transient assays, 0.01 µg of Renilla Luc plasmid was included to normalize transfection efficiency. Dual Luc assays were performed using Dual-Luciferase® Reporter Assay system (Promega, E1901).Fundus fluorescein angiography (FFA)
Fluorescein angiography was performed using a Heidelberg retina angiograph (HRA) system. P18 mice were anesthetized with an intraperitoneal (IP) injection of 1% soluble sodium pentobarbital (40 mg/kg), and the pupils were dilated by 0.1%tropicamide solution(Santen, Osaka, Japan). Fluorescein images were captured at different time points after IP injection of 0.1 ml of 2% fluorescein sodium (Novartis).Statistical analyses
All data were presented as mean ± SD or mean ± SEM. Statistical analysis was performed using the GraphPad Prism software (GraphPad Prism Software, Inc., San Diego, CA, USA). The results were analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni correction for multiple comparisons and student’s t-test for comparisons between two groups.RESULTS
p107 suppresses retinal angiogenesis in Vhl null retina
The genotyping images are shown in Figure 1A (p107), 1B (Vhl) and 1C (α-Cre). We first examined the retinal blood vessels of live animals in FFA. While p107 -/-mice had normal retinal blood vessels (Figure 1D), both Vhl -/- and p107-/-Vhl -/- DKO mice showed delayed hyaloid vessels, which blocked the fluorescein signal in the retina (Figure 1D). Retinal section (Figure 1E) and whole mount (Figure 1F) IB4 staining also proved p107-/-mice had no changes in retinal angiogenesis (Figure 1G). Vhl-/- retina had reduced retinal angiogenesis with a few vessels invading the outer nuclear layer (ONL) (Figure 1E, arrow). Still, the branch points in all three retinal plexus were reduced (Figure 1F-G).Figure 1 Retinal angiogenesis in the p107/Vhl DKO retinas
A-B: The gel images of genotyping of mice of p107-/- (A), Vhl floxed (B) and α-Cre (C). D. FFA images of the P18 eyes of indicated genotypes. E. Horizontal P18 retinal sections of indicated genotypes were stained for nuclear (DAPI, blue) and vascular endothelium cells (IB4, green). F. IB4 staining of P18 wholemount retinas of indicated genotypes. Blow-up shows the vascular density of IVP. G. Quantification of vessel branching points per fields of view [FOV] by AngioTool software. Error bars represent SD of measurements from three retinas (n=3) and asterisks indicate significant differences between WT and indicated genotypes (*P<0.05, **P<0.01, one-way ANOVA followed by Bonferroni correction). The dotted lines in C indicate the boundary between no Cre expression (in the center) and alpha-Cre expression areas (in the periphery). ONL: outer nuclear layer; INL: inner nuclear layer; IVP: intermediate vascular plexus; GCL: ganglion cell layer. Scale bar is 50 µm in E; 200 µm in F.
On the other hand, in the p107/VhlDKO retinas, there were many new blood vessels in the peripheral retina (Figure 1D-E). These vessels penetrated all retinal layers to form a dense capillary bed, and there was no normal SVP (superficial vascular plexus), IVP (intermediate vascular plexus), or DVP (deep vascular plexus) (Figure 1F). To compare the vascular density to other genotypes, we used vascular images from similar depths below the GCL (ganglion cell layer) to represent the IVP and DVP for the DKO retina. The branch points were 2-3 times higher than in WT, p107KO, and VhlKO retina (Figure 1G). Thus, deleting p107 unleashes the proangiogenic effects of Vhl loss in the retina.
p107 affects photoreceptors and glial cells in the Vhl null retina
We also looked at the effects of p107 loss on other retinal cells in Vhl null retinas by staining the P18 p107/VhlDKO retina with cell type-specific markers. While we didn’t find any effects on two excitatory neurons including ganglion cells (Brn3+) and bipolar cells (Chx10+), two inhibitory neurons including amacrine cells (Ap2a+) and horizontal cells (Onecut2+or OC2+), p107 loss significantly reduced rod and cones (Figure 2A-B), and increased Sox9+Müller cells and IBA1+ microglia cells in VhlKO retina (Figure 2A-B). GFAP staining indicated that p107 loss activated Müller cells in Vhl null retinas (Figure 2A-B).p107 affects transcriptome of the Vhl null retina
To further understand the vascular and cellular phenotypes in the p107/Vhl DKO, we run RNA sequencing on p14 WT, p107KO, VhlKO, and p107/Vhl DKO retinas. We analyzed differential expression genes (DEGs) and identified 144 p107KO-related DEGs, 1918 VhlKO-related DEGs, and 2874 p107/Vhl DKO-related DEGs compared to WT (Figure 4A). Gene list enrichment found 3, 49, and 125 pathways in p107KO, VhlKO, and p107/Vhl DKO retinas (Figure 4B). For DKO retinas, the most enriched up-regulated DEGs were in the pathways of cancer, focal adhesion, ECM-receptor interaction, Hif-1, PI3K-Akt, and many inflammatory pathways (Figure 4C), the most enriched down-regulated DEGs were in the pathway of phototransduction (Figure 4D). These changes were readily observed by the heatmap (Figure 4E), especially the changes in the Hif-1 pathway presented in the heatmap (Figure 4F). These RNA sequencing data support the notion that p107 constrains the induction of Hif1 targets in the absence of Vhl.p107 binds to the promoter and inhibits the transcription of Hif target genes
Deleting p107 in Vhl -/-retina increased the expression of several Hif targets, such as Epo, Tek, VEGFA, and Kdr, about 1.7 to 2.2-fold (Figure5A). We ran ChIP assays to test whether p107 directly regulates these genes in the retina. It was challenging to obtain enough pure knockout material from the peripheral Cre-positive region of the α-Cre; Vhlf/f retinas; thus, we assessed p107 binding in WT retinal explants left untreated or exposed to CoCl2, which mimics hypoxia and induced Hif-1αaccumulation (Figure 5B).[29] P107 was bound to the promoters of several Hif targets such as Epo, Kdr, Tek and Vegfa (Figure 5C). P107 can bind these promoters without CoCl2 treatment, indicating that p107 binds E2f, which was recruited to the promoter of these targets. However, CoCl2 treatment significantly increased p107 binding (Figure 5C), suggesting that Hif-1α might directly recruit p107 to these targets. Thus, it is possible that E2f and Hif-1α bind the promoter of these target genes, which recruits p107 that inhibit Hif protein or HRE (Hif responsive element).[30]
Figure 5 P107 protein can bind the promoter and suppress the transcription of several Hif target genes
(A). The relative mRNA levels of indicated genes and indicated genotypes were analyzed by RT-PCR. (B). Representative Western blots of Hif-1a proteins in retinal explants of indicated treatments. (C). ChIP using p107 antibody at the promoter of indicated genes and treatments in WT retinal explant cultured from P8 to P11. The enrichment of p107 was quantified using qPCR and normalized to input. (D). The four Rep4-luciferase constructs used in this study. Red triangles indicate E2f binding sites. (E). Representative Western blots of p107 proteins in 293T cells transfected with pMXIE empty vector or pMXIE-p107 construct. (F). Luciferase reporter assay. 293T cells were transfected with the indicated luciferase-reporter plasmid, pMXIE empty vector, or pMXIE-p107 construct by Lipofectamine. Renilla Luciferase plasmid was included to normalize for transfection efficiency. Error bars represent SEM (in A) or SD (in C, F) of measurements from three retinas (A, C; n=3) or three assays (F; n=3) and asterisks indicate significant differences between WT and indicated genotypes in A; or between IgG No CoCl2 treatment and other indicated treatments in C, between pMXIE and pMXIE-P107 in F (*P<0.05, **P<0.01, one-way ANOVA followed by Bonferroni correction).
Together, the above genetic and molecular analyses reveal that p107 inhibits a subset of Hif1 target genes, providing a logical mechanism to explain why the Vhl -/-retina does not exhibit vessel overgrowth phenotypes, such as RAP or RCH.
DISCUSSION
The Vhl-/-mouse retina up-regulates Hif. However, it does not induce retinal vascular overgrowth, such as MNV3-like lesions or RCH. It is unclear how Hif target expression is suppressed in this context. We previously found that Rb1 can bind and suppress Hif target genes in the Vhl -/-mouse retina. There are E2f binding sites in many Hif target genes,[32] likely, E2f recruits Rb1 to these promoters to inhibit their expression. While Rb1/Vhl DKO retina shows extensive retinal neovascularization, there is still no MNV3-like lesion or RCH, except triple knockout Rb1/p107/Vhl together.[22] Thus, p107 is essential for forming subretinal angiomatous lesions in this context. Our current study indicated that, like Rb1, p107 is also a critical angiogenic inhibitor. Thus, the p107/VhlDKO retina causes widespread retinal neovascularization. As the p107KO mouse retina has no detective phenotype, it is not likely to be an indirect consequence of other cellular effects of p107 loss. Even though we only tested three Hif target genes (Vegfa, Kdr, and Tek genes) in the luciferase assay, RNA sequencing revealed that p107 suppressed many more targets in the Hif pathway of the Vhl -/-mouse retina.While there is no evidence that Hif-1α can recruit Rb1 to the promoter of these Hif target genes,[22] our CHIP data supports the notion that Hif-1α can recruit p107 as CoCl2 treatment greatly enhanced the p107 binding to the promoter of Hif targets. It is well known that Rb1 preferentially interacts with E2f1-3, and p107 mainly binds to E2f4/5. P107 and E2f4/5 are both components of the DREAM complex(Dp, Rb-like, E2F4/5, and MuvB), which is a transcriptional repressor.[33]FoxM1, one component of the DREAM complex, is a downstream target of Hif-1α.[34] Thus, it is possible that Hif-1α recruit p107 through the DREAM complex.
Together, these data present a strong case that p107 directly inhibits Hif target expression in the retina and explains why vascular growth is constrained in the Vhl -/-murine tissue. Our work exposes previously unrecognized roles for the pocket protein family (Rb1 and p107) in constraining retinal angiogenesis, which will be essential to dissect the pathogenesis of MNV3 and RCH.
Besides our Rb/p107/VhlTKO mouse model, many studies use other Cre strains to knock out the Vhl gene in the mouse eye to study the mechanism of MNV3, RCH, or retinal neovascularization. The significant differences between our model and these models are in three aspects: different targeting cells, knockout strategies (single VhlKO or combined VhlKO with other gene knockouts), and potential cell-of-origin of these lesions. For instance, removing Vhl in murine hemangioblasts using Scl-CreER triggered many features of early-stage RCH, including dilated tortuous vessels, vascular leakage and foamy stromal cell clusters.[35] This mouse model is a single VhlKO; the knockout occurs in hemangioblasts, which can differentiate into vascular endothelium cells, and it suggests that embryonically arrested hemangioblasts are the cell-of-origin of RCH. In addition, this model can produce hemangioblastoma-like lesions in the mouse brainstem, like human VHL diseases.[35] This work is a milestone in the mouse RCH model field, but the lesions are relatively minor. Pathologically, they only present some “tumorlet” cells adjacent to the dilated vessels. It will be exciting to examine the expression of pocket proteins in these hemangioblasts and test if removing Rb1 or p107 can exaggerate their phenotypes.
Pde6g-CreERT2-mediated VhlKO in rod cells can cause retinal neovascularization, resembling the early stage of human MNV3.[35] However, PAS staining only identified very few subretinal new vessels. Thus, this model differs significantly from ours regarding the subretinal vascular phenotypes.[22] This mouse model is like other well-known models for early stage of MNV3, such as VhlKO in mouse RPE cells,[36] and models related to abnormal lipid metabolism (Vldlr-/- mice,[37] Cyp27a1-/- mice[38]), or abnormal cell migration pathway (endothelial cell-specific Srf knockout mice[39]), and RNV3 (JR5558) mice which have mutations in genes regulating retinal polarity and inflammation.[19,40]
In the mouse model of oxygen-induced retinopathy (OIR), LysmCre-mediated VhlKO in myeloid cells can promote the expression of VEGF and bFGF, enhance retinal vascular regeneration, and reduce pathological retinal neovascularization.[41] This model lacks MNV3/RCH-like lesions but reveals a complex interaction between myeloid cells and vascular endothelium cells under hypoxia. However, this work suggests that myeloid cells may not be the cells of origin of RCH, as myeloid VhlKO reduces pathological retinal neovascularization.
In summary, our MNV3/RCH-like mouse model is unique in three aspects. First, it is mainly based on the Pax6 α-Cre mediated VhlKO in retinal progenitors. Second, it knocked out the other two genes (Rb1 and p107) and the Vhl gene.[22] Similarly, Vhl loss alone does not induce kidney Vhl disease, but Rb1/p53/Vhl triple knockout induces a CCRCC (clear cell renal cell carcinoma) mouse model.[42] Based on these two works, it seems likely that while VHL mutation alone can induce VHL disease in humans (including RCH and CCRCC), it can’t cause VHL diseases in mice. Third, we show that the foamy cells in the MNV3/RCH-like lesions originate from Müller glia, which express high levels of Sox9 and Gal3. This is consistent with a report that Gal3 is a diagnostic marker for human brain hemangioblastoma,[43]which indicates that Gal3 is also a possible biomarker for diagnosing human RCH.[22]
Correction notice
NoneAcknowledgement
We thank P Gruss and M Rudnicki for the mice, K Zhao and R Bremner for the plasmid vector, and T Yu and S Lu for the luciferase reporter assay. This work was supported by grant to DC from the National Natural Science Foundation of China (82171063). DC was the recipient of the Eugene Chan Award in 1995. We want to dedicate this manuscript to the memory of Professor Eugene Chan for his 125th anniversary of birth and to the celebration of his tremendous contribution to the development of Ophthalmology at West China Hospital (previously Cun-ren Hospital in Chengdu).Author Contributions
(Ⅰ) Conception and design: Danian Chen(Ⅱ) Administrative support: Lirong Xiao
(Ⅲ) Provision of study materials or patients: Danian Chen
(Ⅳ) Collection and assembly of data: Ling Huang, Yiwen Hong,Wenyu Du, Wei Qiang, Wenyue Chen, Yujiao Wang, Ran Wei
(Ⅴ) Data analysis and interpretation: Ling Huang, Yiwen Hong, Wenyu Du, Danian Chen
(Ⅵ) Manuscript writing: All authors
(Ⅶ) Final approval of manuscript: All authors