铁死亡与眼表疾病的关系研究进展
'%20fill='white'%20fill-opacity='0.01'/%3e%3cmask%20id='mask0_3477_29692'%20style='mask-type:luminance'%20maskUnits='userSpaceOnUse'%20x='0'%20y='0'%20width='16'%20height='16'%3e%3crect%20id='&%23232;&%23146;&%23153;&%23231;&%23137;&%23136;_2'%20x='16'%20width='16'%20height='16'%20transform='rotate(90%2016%200)'%20fill='white'/%3e%3c/mask%3e%3cg%20mask='url(%23mask0_3477_29692)'%3e%3cpath%20id='&%23232;&%23183;&%23175;&%23229;&%23190;&%23132;'%20d='M14%205L8%2011L2%205'%20stroke='%23333333'%20stroke-width='1.5'%20stroke-linecap='round'%20stroke-linejoin='round'/%3e%3c/g%3e%3c/g%3e%3c/svg%3e)
关键词
摘要
铁死亡作为一种独特的细胞死亡方式,近年来在眼科疾病研究中不断被提及,其重要性不容忽视。该过程涉及铁依赖的脂质过氧化,导致细胞膜结构受损,最终引发细胞死亡。在眼科领域,铁死亡与多种退行性疾病的病理进程紧密相连,为这些疾病的治疗提供了新的视角。在视网膜退行性疾病中,铁死亡通过影响视网膜细胞的生存与死亡平衡,加速疾病进程,这一发现为视网膜退行性疾病的治疗提供了新的靶点和策略。值得关注的是,研究表明铁死亡在眼表相关疾病的发生、发展过程中也扮演重要角色,随着关于铁死亡调节机制的研究进展,以及针对铁死亡过程的治疗策略和方法的发展,铁死亡在眼表疾病中的作用及其潜在的治疗价值逐渐被揭示。本文着重介绍了4条铁死亡常见信号通路在铁死亡过程中发挥的作用:System Xc−/GSH/GPX4通路通过调控谷胱甘肽合成与抗氧化活性抑制铁死亡;FSP1/CoQ10/NAD(P)H通路以独立于GPX4的方式中和脂质过氧化自由基;Nrf2/HO-1通路通过调控氧化还原稳态及铁代谢相关基因表达发挥保护作用;GCH1/BH4通路则通过调节四氢生物蝶呤合成参与铁死亡调控。综述了铁死亡内部机制及其与炎症的相互作用,并总结了目前铁死亡在眼表疾病中的研究进展,为开发眼表角膜受损的新治疗策略提供见解。
全文
文章亮点
1 关键发现
• 系统阐明4条铁死亡关键信号通路(System Xc−/GSH/GPX4、FSP1/CoQ10/NAD (P) H、Nrf2/HO-1、GCH1/BH4)的分子机制,及其与炎症反应的双向调控关系:铁死亡可通过脂质过氧化产物激活炎症通路,炎症因子又可反过来抑制抗氧化通路、促进铁积累,形成恶性循环。该过程与角膜碱烧伤、干眼、角膜炎等疾病的发生发展直接相关,且在角膜损伤愈合中发挥关键作用。
2 已知与发现
• 系统串联4条核心信号通路,揭示其在眼表细胞铁死亡中的协同调控网络,明确各通路在维持氧化还原稳态、抑制脂质过氧化中的特异性作用;阐明铁死亡与眼表疾病中炎症、新生血管形成、纤维化的分子关联。证实铁死亡抑制剂不仅能保护眼表细胞,还可通过调控炎症—铁死亡恶性循环、靶向病原体铁死亡等多维度发挥治疗作用,拓展了铁死亡抑制剂的应用场景。
3 意义与改变
• 建立了铁死亡与眼表疾病病理机制的完整关联框架,深化了对眼表疾病“铁代谢—氧化应激—炎症”调控网络的理解,为眼表疾病的发病机制研究提供了全新的分子视角。为后续开展眼科疾病精准治疗和铁死亡相关信号通路抑制剂的筛选、临床转化研究提供了新的视角。
角膜作为眼睛最外层的透明组织,含有丰富的神经末梢,对疼痛、温度、触觉十分敏感,不仅是眼睛的主要屈光介质之一,而且具有一定保护眼球内部结构免受外界物理和化学伤害的作用。当角膜受到损伤时,便会从原有的血管结构中萌发出新生血管,如来自角膜周围神经丛的毛细血管和小静脉[1],随后是免疫细胞的浸润和炎症介质的释放,在炎症反应之后,角膜上皮细胞及基质中的成纤维细胞开始增殖,以覆盖损伤区域,产生新的胶原纤维,修复受损的基质层,角膜得以重塑,以恢复其正常的结构和功能[2-3]。如果角膜愈合过程中出现感染或过度的炎症反应,可能会导致角膜瘢痕形成,从而影响角膜的透明性和视力。近期研究发现,铁死亡在角膜损伤及愈合过程中发挥着重要作用,铁死亡与炎症反应与受损角膜的愈合密切相关。
铁死亡是一种不同于自噬、焦亡等细胞死亡形式的由铁驱动脂质过氧化(lipid peroxidation, LPO)介导的程序性细胞死亡过程[4],细胞内铁代谢的失调导致细胞内铁离子的积累催化产生活性氧(reactive oxygen species, ROS)可导致脂质过氧化的增加,进而引发细胞死亡。铁死亡与多种疾病过程有关,如癌症、缺血再灌注损伤(ischemia-reperfusion injury, IRI)、神经退行性疾病、炎症性疾病、脑损伤等[5]。铁死亡的分子机制包括谷氨酸/胱氨酸抗转运体(System Xc−)、抗氧化系统、铁代谢、不饱和脂肪酸代谢、铁沉抑制蛋白1-泛醌系统 (ferroptosis suppressor protein 1-coenzyme Q system, FSP1-CoQ)等。本文重点研究了胱氨酸 - 谷氨酸反向转运体系统 Xc−/谷胱甘肽/谷胱甘肽过氧化物酶4 (cystine/glutamate antiporter system Xc− / glutathione / glutathione peroxidase 4, System Xc− / GSH/GPX4)、铁死亡抑制蛋白1/辅酶Q10/还原型烟酰胺腺嘌呤二核苷酸(磷酸) [ferroptosis suppressor protein 1 / coenzyme Q10 / nicotinamide adenine dinucleotide (phosphate) hydrogen, FSP1 / CoQ10 / NAD(P)H]、核因子红系2相关因子2/血红素氧合酶-1 (nuclear factor erythroid 2-related factor 2/heme oxygenase-1, Nrf2/HO-1)、GTP 环水解酶1/四氢生物蝶呤 (GTP cyclohydrolase 1/tetrahydrobiopterin, GCH1/BH4)通路(图1),阐述了它们的发生机制,并讨论了它们与炎症的相互作用,回顾了眼表疾病中铁死亡相关研究。
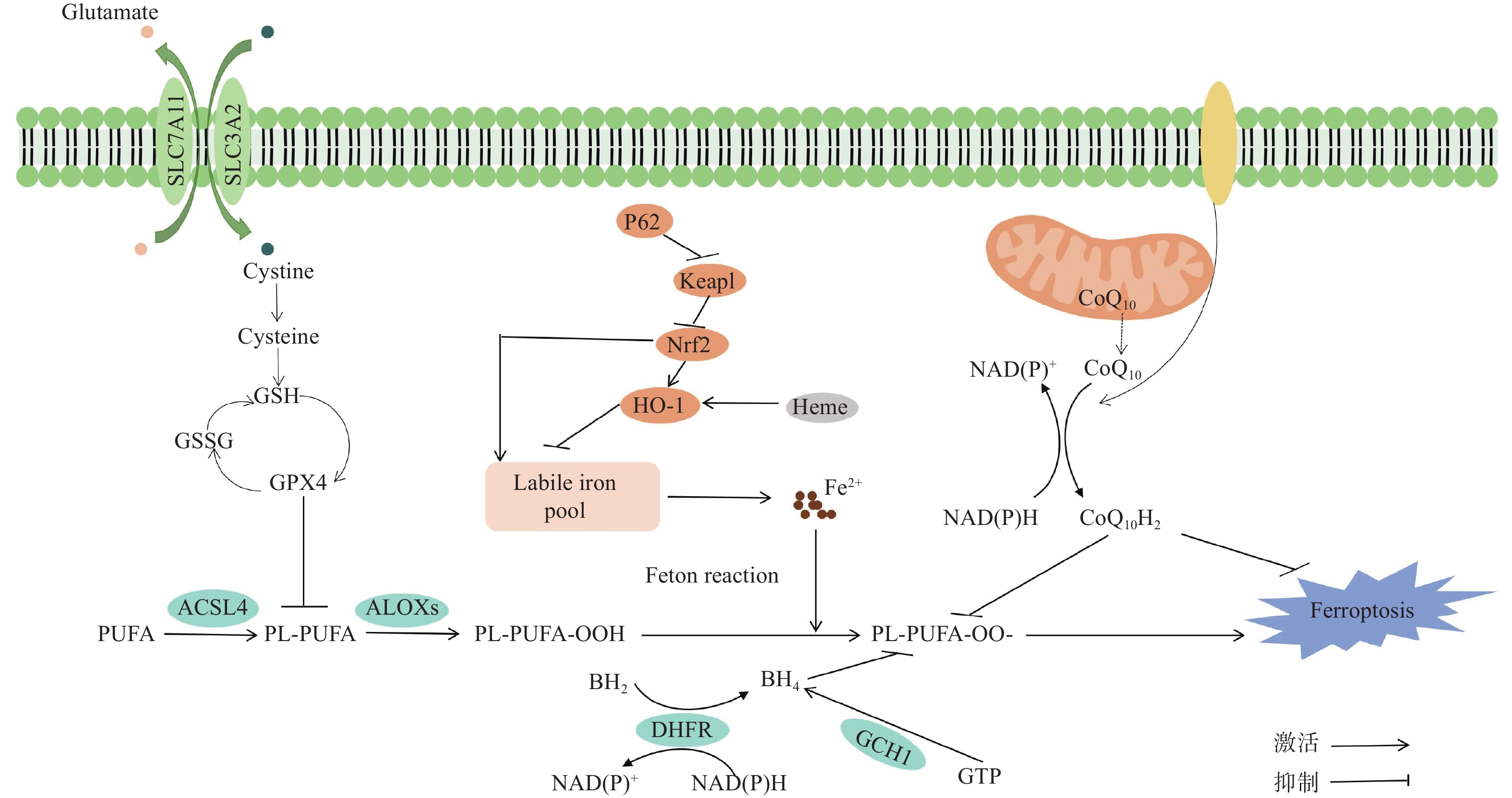
1 铁死亡的常见信号通路
1.1 System Xc−/GSH/GPX4通路
谷胱甘肽(glutathione, GSH)是一种重要的抗氧化剂,能够清除细胞内的自由基,保护许多蛋白质和酶等分子中的巯基不被有害物质氧化,从而保证蛋白质和酶等分子生理功能的正常发挥[6];谷胱甘肽过氧化物酶4 (glutathione peroxidase 4, GPX4)是一种关键的硒酶,能够利用GSH将有毒的脂质氢过氧化物转化为无毒的脂质醇来防止铁死亡[7]。胱氨酸/谷氨酸反向转运体System Xc−位于细胞膜上,是由溶质载体家族7成员11 (solute carrier family 7 member 11, SLC7A11)和溶质载体家族3成员2 (solute carrier family 3 member 2, SLC3A2)两个亚基通过二硫键连接而成的异二聚体复合物[7],SLC7A11是一个主要的功能亚基,可以将胱氨酸转运到细胞中合成GSH[8],这一过程可以调节GSH的生成及GPX4的释放,从而调节细胞氧化还原稳态。Dixon等[9]研究证明,铁死亡诱导剂Erastin可以通过抑制System Xc−来消耗GSH,从而阻碍胱氨酸的摄取。同样的,肿瘤抑制蛋白p53的下调可以使SLC7A11的表达减少抑制GSH的合成,从而增加ROS诱导的铁死亡的敏感性[10],补充GSH可通过调节SLC7A11/GPX4轴抑制铁死亡[11]。System Xc−/GSH/GPX4通路还可以通过调节炎症细胞因子的产生来影响疾病的发生、发展过程。当此通路受损时,细胞内GSH水平降低,GPX4 活性受到抑制,导致氧化应激增加,进而激活核因子κB (nuclear factor-kappa B, NF-κB)等转录因子[12],NF-κB的激活会促进多种炎症细胞因子的基因转录,如白细胞介素-6 (interleukin-6, IL-6)、肿瘤坏死因子-α (tumor necrosis factor-α, TNF-α)和IL-1β等,这些细胞因子会进一步加剧炎症反应;当该通路正常运作时,可以有效减少丙二醛(malondialdehyde, MDA)和4-羟基壬烯酸(4-hydroxynonenal, 4-HNE)等脂质过氧化产物的生成[12-13]。在炎症状态下,产生的ROS及促炎细胞因子等多种炎症介质,会对 System Xc−/GSH/GPX4通路产生抑制作用。ROS可以氧化修饰System Xc−的亚基,影响其转运胱氨酸的功能,导致胞内胱氨酸摄取减少,GSH合成受限,加剧细胞与组织中铁死亡的发生[14]。有研究表明,过量的TNF-α累积能激活NF-κB通路,NF-κB入核后可调控一系列基因表达,其中部分基因产物又能够反作用于System Xc−可能干扰 SLC7A11的正常功能,阻碍胱氨酸摄取,进而减少GSH合成[15]。此外,干扰素-γ (interferon-gamma, IFN-γ)作为一种重要的免疫细胞因子,能够通过激活Janus激酶-信号转导与转录激活因子(Janus kinase-signal transducer and activator of transcription, JAK-STAT)信号通路,抑制System Xc−的关键组分SLC7A11的表达,导致GSH合成减少[16]。GSH含量降低会削弱GPX4的活性,使得细胞无法有效清除脂质氢过氧化物,引发铁死亡。同时,铁死亡过程中产生的脂质过氧化产物又可作为炎症信号分子,进一步激活炎症细胞,释放更多炎症因子,形成炎症与铁死亡的恶性循环。System Xc−在抗氧化中起关键作用,可通过调节GSH的产生和GPX4的激活来有效抑制铁死亡,同时,System Xc−/GSH/GPX4 通路又可以通过降低炎症相关信号通路的激活,减轻炎症对细胞和组织的损伤。
1.2 FSP1/CoQ10/NAD(P)H通路
铁死亡抑制蛋白(ferroptosis suppressor protein 1, FSP1),是一种独立于GPX4起作用的铁死亡抑制剂[17],能够自动定位到质膜中并中和脂质过氧化自由基。二氢烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate, NADPH)是细胞还原能力的主要来源[18],NADPH利用泛醌(coenzyme Q10, CoQ10)作为抗氧化剂捕获自由基,FSP1利用NADPH促进CoQ10还原为活性形式,从而防止磷脂过氧化[19-20]。CoQ10是一种主要存在于线粒体内膜中的亲脂性分子,作为一种亲脂性抗氧化剂能够捕捉自由基以防止脂质过氧化,还能间接再生另一种抗氧化剂α-生育酚以捕捉自由基,从而抑制铁死亡[21]。近年研究强调了FSP1在调节细胞铁死亡中的重要性,该研究发现FSP1的选择性有效抑制剂(ferroptosis suppressor protein 1 inhibitor, IFSP1)可抑制细胞中FSP1的表达,从而诱导铁死亡,减少肿瘤的发生[22]。在视网膜色素上皮(retinal pigment epithelium, RPE)细胞中,过表达FSP1还能显著下调 CoQ10/NAD(P)H 和脂质过氧化反应,有效减少RPE细胞中铁死亡的发生[23]。FSP1/CoQ10/NAD(P)H通路在功能上独立于GPX4,但与GPX4及GSH相互协同,对抑制细胞脂质过氧化及铁死亡有一定的作用[24]。而当FSP1功能受限时,CoQ10还原为活性形式受阻,无法有效捕获自由基,细胞脂质过氧化水平升高,使铁死亡的易感性增加。反过来,铁死亡发生时,释放的细胞内容物可激活炎症小体,如NOD样受体家族含pyrin结构域蛋白3 (NOD-like receptor family pyrin domain containing 3, NLRP3)炎症小体,促使IL-1β、IL-18等炎症因子成熟并释放,加剧炎症反应[21]。同样地,炎症反应也会干扰FSP1/CoQ10/NAD (P) H通路的正常功能。当丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)信号通路激活后,可通过抑制FSP1的启动子活性、促进FSP1蛋白的泛素化降解、促进FSP1基因启动子区域的组蛋白去乙酰化的方式来抑制FSP1的表达与活性,导致CoQ10H2水平下降,脂质过氧化增加,从而促进铁死亡[25]。炎症期间,产生的大量ROS可氧化修饰FSP1,使其结构和功能改变,影响其定位到质膜中和脂质过氧化自由基的能力。此外,炎症介质还可能干扰NAD (P) H的生成或利用,从而影响该通路的正常功能,从而加剧铁死亡的发生[26]。
1.3 Nrf2/HO-1通路
核因子红细胞2相关因子2 (nuclear factor E2-related factor 2, Nrf2)是一种对氧化应激高度敏感的转录因子,不仅可以调控细胞氧化还原稳定性,还可以通过靶向GSH合成和铁代谢相关基因的表达,保护细胞免于铁死亡[27]。Nrf2已被鉴定有多个靶基因,如血红素加氧酶1 (heme Oxygenase 1, HO-1)、SLC7A11、铁蛋白轻链(ferritin light chain, FTL)和GPX4等[11, 27]。其中,HO-1还参与炎症反应、组织损伤、细胞死亡的抑制过程。关于Nrf2/HO-1研究越来越多,Zhao等[28]研究表明,双硫仑/铜复合物可以抑制Nrf2/HO-1通路,增加细胞中游离Fe2+,增强脂质过氧化,从而促进铁死亡。Choi等[29]发现HO-1短暂的低表达可以促进氧化还原活性游离铁的捕获,通过诱导铁蛋白(一种内源性铁清除剂)和线粒体生物生成避免芬顿(fenton)反应和ROS的产生,同时在这一阶段HO-1与Nrf2之间的相互作用可能会促进GPX4的生成,从而抑制铁死亡的发生,反之HO-1 的长期高表达可促进细胞铁积累,从而诱发活性氧ROS产生、炎症、线粒体功能障碍和铁死亡。同样地,适度的炎症刺激也可以作为一种应激信号,激活Nrf2/HO-1通路。当细胞受到炎症因子、细菌毒素或其他炎症刺激时,细胞内的氧化还原状态发生改变,产生一定量的ROS,这些物质可以作为信号分子,激活Nrf2/HO-1通路。这种激活是机体的一种自我保护机制,旨在增强细胞的抗氧化能力,抵御炎症损伤。在炎症初期,细胞受到氧化应激刺激,Nrf2被激活并转位至细胞核,与包括HO-1在内的抗氧化反应元件(antioxidant response element, ARE)结合,启动一系列抗氧化基因表达。HO-1可催化血红素分解,产生具有抗炎、抗氧化作用的一氧化碳、胆绿素等物质,抑制炎症反应[30]。然而,在慢性炎症状态下,炎症信号通路持续激活,如NF-κB通路过度活化,会抑制Nrf2的活性,减少HO-1表达。同时,炎症产生的ROS可氧化修饰Nrf2,使其与Keap1结合更紧密,被蛋白酶体降解,导致细胞抗氧化能力下降,对铁死亡的敏感性增加[31]。而铁死亡发生时,细胞内游离铁离子增多,又会促进炎症反应,形成复杂的相互作用网络。
1.4 GCH1/BH4信号通路
四氢生物蝶呤 (tetrahydrobiopterin, BH4)是抗氧化系统的一个组成部分,参与一氧化氮、神经递质和芳香氨基酸的代谢[32],GTP 环氢酶 1 (GTP cyclohydrolase 1, GCH1) 作为BH4合成的限速酶,当其与GCH1反馈调节蛋白(GCH1-feedback regulatory protein, GFRP)结合时,GCH1活性被苯丙氨酸和BH4水平调节而发生改变,苯丙氨酸与复合物结合并激活GCH1,而BH4与复合物结合抑制GCH1活性[33-34]。Kraft等[35]在2020年使用人类全基因组文库进行CRISPR/dCas9过表达筛选,结果显示GCH1为最显著抑制铁死亡的保护基因,且后续研究证明细胞GCH1水平的增加后,叶酸网络中的下游产物BH4/BH2,被选择性地和强烈地升高,抑制脂质过氧化和铁致细胞死亡。BH4可以调节铁转运蛋白的表达,从而影响细胞对铁的摄取[36-38]。同时,BH4还可以作为抗氧化剂,直接或间接参与清除细胞内的ROS,抑制脂质过氧化,进而调控铁死亡的发生[39-40]。与上述其他通路相似,GCH1/BH4信号通路与炎症反应有着复杂且紧密的相互作用。炎症反应中的ROS增加氧化应激,消耗BH4,导致一氧化氮合酶(nitric oxide synthases, NOS)解偶联,进一步增加ROS,促进铁死亡[41]。而当 GCH1/BH4 通路因铁死亡诱导等被激活时,生成的BH4可促进一氧化氮(nitric oxide, NO)生成[42],阻碍NF-κB激活,减少炎症因子转录释放,抑制炎症反应。但如果铁死亡被过度激活,大量脂质过氧化产物又能够激活NF-κB,加重炎症,形成恶性循环。
2 铁死亡在眼表疾病中的研究
角膜作为重要屈光介质,患者预后视力与治疗后角膜透明程度密切相关,研究发现铁死亡及炎症反应在受损角膜愈合过程中发挥重要作用。因此,角膜损伤的治疗和管理需要综合考虑,以达到最佳的愈合结果。
2.1 铁死亡与角膜碱烧伤
Wang等[43]研究证实在碱烧伤角膜中存在铁死亡这一新型细胞死亡方式,严重损伤会导致ROS相关的谷氨酸 - 半胱氨酸连接酶修饰亚基(glutamate-cysteine ligase modifier subunit, GCLM)、谷胱甘肽过氧化物酶1 (glutathione peroxidase 1, GPX1)、谷胱甘肽二硫化物还原酶(glutathione - disulfide reductase, GSR)、过氧化物氧还酶1 (peroxiredoxin 1, PRDX1)、超氧化物歧化酶1 (superoxide dismutase 1, SOD1)基因mRNA表达水平显著上调,表明碱烧伤后角膜中ROS积累,从而促进脂质过氧化和线粒体结构和功能的破坏;同时,发现碱烧伤角膜中铁死亡关键标志基因前列腺素内过氧化物合酶2 (prostaglandin-endoperoxide synthase 2, PTGS2)的mRNA表达水平、多不饱和脂肪酸氧化反应后的脂质过氧化产物4-羟基壬烯酸(4-hydroxynonenal, 4-HNE)及脂质代谢的重要调节蛋白长链酰基辅酶A合酶4 (acyl-CoA synthetase long chain family member 4, Acsl4)的蛋白表达水平显著升高,铁死亡调节因子GPX4的蛋白表达水平降低,通过特异性抑制剂铁抑素-1 (ferrostatin-1, Fer-1)靶向碱烧伤角膜的铁死亡,观察到部分线粒体结构被恢复、GPX4的表达较前有所提高、脂质过氧化产物及ROS的产生减少,此外,在Fer-1的作用下碱烧伤角膜中IL-1β、IL-6、IL-10炎症因子水平、TNF-α的表达及新生血管相关因子血管内皮生长因子A (vascular endothelial growth factor A, VEGFA)、VEGFC、血管内皮生长因子受体3 (vascular endothelial growth factor receptor 3, VEGFR3)和基质金属蛋白酶2 (matrix metalloproteinase 2, MMP2)的mRNA水平显著降低[44]。这些研究结果表明,在碱烧伤角膜中ROS的累积,导致铁死亡的发生,加重炎症及新生血管的生成,而Fer-1可以抑制铁死亡,保护角膜细胞免受损伤和血管生成,从而促进角膜愈合过程。同样地,Fer-1已被证明在暴露于香烟烟雾和加热烟草制品后能有效地减少细胞死亡和提高细胞活力[45],另一种铁死亡抑制剂UAMC3203也被发现在体内和体外通过刺激细胞迁移来加速角膜上皮伤口愈合[46]。以上结果表明,铁死亡在受损角膜的愈合过程中扮演重要角色,铁死亡抑制剂能够通过不同作用机制,在细胞损伤和组织修复过程中发挥保护与促进作用。
2.2 铁死亡与干眼症
研究者在干眼模型中发现,醛酮还原酶家族1成员C1 (aldo-keto reductase family 1 member C1, AKR1C1)作为依赖于NAD(P)(H)催化羰基底物还原的超家族蛋白亚家族中成员,被抑制后其将脂质过氧化物酶的毒性副产物4-HNE转化为无毒的1,4-二羟基-2壬烯的能力显著降低,从而促进角膜上皮细胞 (human corneal epithelial cells, HCECs)铁死亡的发展,导致随后的炎症反应和干眼症,而Nrf2的激活能够显著上调高渗条件下HCECs中AKR1C1的表达,AKR1C1的上调能够保护角膜上皮细胞免受脂质过氧化损伤,减少细胞铁死亡的发生,减轻干眼症状[47]。Hou等[48]研究表明,虾青素 (astaxanthin, AST)在小鼠干眼模型中能够上调SLC7A11和GPX4,抑制铁死亡;在高渗诱导的干眼上皮细胞中,AST能够增加转铁蛋白的表达,促进铁储存,减少Fe2+过载,增加GSH和GPX4,清除ROS和过氧化脂质,挽救线粒体结构,防止铁死亡。同样的,Dai等[49]在苯扎氯铵诱导的大鼠干眼模型中发现,盘状蛋白结构域受体 (disidin domain receptor, DDR)表达升高,而特异性的DDR-1抑制剂(DDR1-IN-1)在体内能够抑制DDR1的表达和磷酸化,降低LPO、铁蛋白重链1 (ferritin heavy chain 1, FTH1)、FTL与ACSL4的表达,减轻ACSL4介导的铁死亡;在高渗刺激的HCSCs中DDR1-IN-1可以通过抑制DDR1,减少干眼模型中ACSL4介导的铁死亡的顺式调节因子 Yes相关蛋白 (Yes-associated protein, YAP)的核累积,减弱Hippo/YAP通路的激活,抑制铁死亡,从而减轻干眼症状。
2.3 铁死亡与角膜炎
Chen等[50]研究表明,由左氧氟沙星联合Fer-1治疗的细菌性角膜炎(bacterial keratitis, BK)小鼠模型中髓过氧化物酶 (myeloperoxidase, MPO)、TNF-α和IFN-γ炎症因子与纤维化因子α-平滑肌肌动蛋白 (α-smooth muscle actin, α-SMA)的表达明显减少,与此同时,左氧氟沙星联合Fer-1治疗组碱烧伤角膜中GPX4、SLC7A11的表达增多,从而减少铁死亡的发生,减轻BK中的炎症和瘢痕形成;体外实验中,Fer-1处理降低了LPS诱导的角膜基质干细胞 (corneal stromal stem cells, CSSCs)中ROS、Fe2+和α-SMA的表达,此外,Fer-1还升高了CSSCs中GPX4和SLC7A11的mRNA表达和蛋白表达。这些结果表明,Fer-1能够通过激活GPX4/SLC7A11轴,减少BK角膜细胞的铁死亡,减轻角膜炎症并促进角膜愈合。除此之外,利用负载FeSO4的水凝胶治疗耐甲氧西林金黄色葡萄球菌(methicillin-resistant S. aureus, MRSA)诱导的小鼠角膜炎模型发现,FeSO4诱导了金黄色葡萄球菌的ROS累积和脂质过氧化,促进其铁死亡的发生,抑制其生物膜形成,最终诱导金黄色葡萄球菌裂解并抑制其分裂,致使小鼠角膜炎快速恢复[51]。这些发现强调了靶向铁死亡在治疗角膜相关疾病的炎症和组织损伤中的潜力。
2.4 铁死亡与其他眼表疾病
角膜作为主要屈光介质之一,在近视的发生、发展过程中发挥重要作用。角膜中铁死亡的过程影响近视的发病进展。Chen等[52]利用定量串联质量标签 (tandem mass tag, TMT)标记法和平行反应监测 (parallel reaction monitoring, PRM)验证高通量蛋白组学分析结果,研究了低度近视与高度近视患者角膜基质中的蛋白表达差异,分析结果表明人近视角膜基质中的铁死亡及缺氧诱导因子1 (hypoxia-inducible factor 1, HIF-1)信号通路被激活,FTL及FTH1与近视的发生、发展呈负相关,转铁蛋白 (transferrin, TF)与近视的发展呈正相关。表明铁死亡与氧化应激与近视发展过程密切相关,但目前铁死亡在近视发展过程中的作用及机制尚不明确,需要进一步通过生物学与功能学实验进行分析与验证。此外,利用GSE204791转录组数据集矩阵进行生物学信息分析后,进一步筛选圆锥角膜 (corneal keratoconus, KC)枢纽基因与铁死亡基因重叠的核心模块,发现在圆锥角膜患者角膜上皮中醛酮还原酶家族 1 成员C3 (aldo-keto reductase family 1 member C3, AKR1C3)其表达减少,AKR1C3具有氧化还原酶活性和AKR (NADPH)活性并参与肿瘤细胞氧化还原稳态和免疫的调节[22, 53],AKR1C3蛋白表达的降低可以诱导铁死亡的发生[54]。AKR1C3表达减少使得负责蛋白水平调控的miRNA-184的合成也受到损害且预测吲哚美辛、柔红霉素、多柔红霉素、多西紫杉醇为靶向has-miR-184减轻KC铁死亡的有效药物[55],不过目前尚未有实验验证分析结果的准确性。
3 小结与展望
角膜作为眼球的前部透明组织,其结构和功能的完整性对于视觉的维持至关重要。然而,角膜易受到各种外界因素的损伤,如化学烧伤、物理性创伤等,这些损伤会破坏角膜组织的完整性,引发炎症反应和新生血管形成,从而影响角膜的透明度和视觉功能。在受损角膜的愈合过程中,铁死亡扮演了重要角色。损伤后的角膜组织中,ROS和游离铁离子的水平显著升高,这些物质会催化细胞膜上的不饱和脂肪酸发生过氧化反应,导致角膜细胞发生铁死亡。铁死亡的发生不仅加剧了角膜组织的损伤程度,还延缓了角膜的愈合过程。通过抑制铁死亡的发生,可以减轻角膜组织的损伤,促进角膜细胞的增殖和分化,从而加速角膜的愈合过程。这一发现对于开发新的角膜损伤治疗方法具有重要意义,有望为角膜损伤患者提供更加有效和安全的治疗方案。
受损角膜修复过程中,不仅有铁死亡的发生还有炎症反应的影响,铁死亡通过多种途径影响炎症反应的进程,炎症反应通过多种机制调节铁死亡的发生和发展。铁死亡过程中,铁离子催化产生的ROS可导致细胞膜脂质的过氧化,进而引发炎症反应。脂质过氧化产物如MDA和4-HNE等可以作为炎症信号分子,激活炎症细胞,促进TNF-α、IL-1β和IL-6等炎症介质的释放[43, 56]。这些炎症因子可进一步激活免疫细胞,导致它们向炎症部位迁移和聚集,释放更多的炎症介质,加剧炎症反应。炎症细胞的浸润和炎症介质的释放可破坏组织结构,影响组织修复和再生。同时在炎症反应中,中性粒细胞、巨噬细胞等免疫细胞被激活,通过NADPH氧化酶和线粒体电子传递链产生大量ROS,ROS是NLRP3炎症小体的重要激活信号之一,而NLRP3激活后又可进一步增加ROS的产生。此外,ROS可氧化修饰Nrf2,使其与Keap1结合更紧密,被蛋白酶体降解,导致细胞抗氧化能力下降,对铁死亡的敏感性增加[8]。炎症过程中释放的细胞因子,可以影响与铁代谢相关基因的表达。炎症信号通路的激活可进一步上调铁代谢相关基因的表达,增加铁离子的摄取和积累,从而促进铁死亡的发生,而炎症反应中产生的ROS和活性氮 (reactive nitrogen species, RNS)可以加剧氧化应激,导致脂质过氧化的增加,加剧氧化应激,间接促进铁死亡的发生[57]。铁死亡与其他细胞死亡方式(如凋亡、坏死等)之间存在复杂的相互作用,在眼表疾病的病理过程中,多种细胞死亡方式可能同时发生。如何在复杂的细胞死亡网络中,精准调控铁死亡进程,使其朝着有利于疾病治疗的方向发展,是接下来研究需要解决的关键问题。
目前,已有一些铁死亡抑制剂在眼表疾病研究和应用中取得一定进展。Wang等[43]研究利用纳米载体,将铁死亡抑制剂包裹其中,实现对眼表疾病部位的精准递送,提高药物在病变组织的浓度,增强治疗效果的同时降低了药物在全身的分布,减少不良反应,从药物递送角度来看,纳米技术的发展为新型铁死亡抑制剂的递送提供了新途径。但仍需要开发更高效、更具特异性且不良反应少的新型铁死亡抑制剂,若开发出能够特异性调节眼表细胞中铁死亡相关通路的抑制剂,就可以在有效抑制铁死亡的同时,减少对其他正常细胞生理功能的影响。此外,由于目前大多数铁死亡相关研究是在细胞模型和动物模型中进行的,从基础研究到临床应用,铁死亡相关研究面临诸多挑战。铁死亡抑制剂的安全性和有效性在临床研究中需要进一步验证。
铁死亡与炎症反应的双向调节机制在眼表疾病的发生、发展中起着关键作用,铁死亡在眼表疾病临床治疗中具有潜在应用价值。新型铁死亡抑制剂的研发为眼科疾病治疗带来了新的希望,进一步理解炎症反应与铁死亡之间的相互作用,对于揭示炎症相关疾病的发病机制和开发新的治疗策略具有重要意义。靶向铁死亡在预防和治疗角膜疾病方面具有很大的前景,为角膜相关疾病的治疗提供了新的思路。
声明
在论文撰写中无使用生成式人工智能。论文撰写中的所有内容均由作者独立完成,并对出版物的真实性和准确性承担全部责任。
利益冲突
所有作者均声明不存在利益冲突。
开放获取声明
本文适用于知识共享许可协议(Creative Commons),允许第三方用户按照署名(BY)-非商业性使用(NC)-禁止演绎(ND)(CC BY-NC-ND)的方式共享,即允许第三方对本刊发表的文章进行复制、发行、展览、表演、放映、广播或通过信息网络向公众传播,但在这些过程中必须保留作者署名、仅限于非商业性目的、不得进行演绎创作。
基金
1. 国家自然科学基金(82060171) 。This work was supported by the National Natural ScienceFoundation of China (82060171).
参考文献
43. 王凯. 铁死亡抑制剂Ferrostatin-1脂质体对碱烧伤角膜的治疗作用的研究[D]. 浙江: 浙江大学, 2021.
Wang K. Functional Study of the Therapeutic Effect of FerroptosisInhibitor Ferrostatin-1 Liposome in the Alkali Burned Corne[D]. Zhejiang: Zhejiang University School of Medicine. 2021.
57. 李建德. 小鼠角膜碱烧伤及雷帕霉素对其修复的机制研究[D]. 兰州: 兰州大学, 2021. DOI: 10.27204/d.cnki.glzhu.2021.000062.
Li JD. The mechanisms of corneal alkali burn and rapamycin-induced damage repair using a mouse model[D]. Lanzhou: Lanzhou University, 2021. DOI: 10.27204/d.cnki.glzhu.2021.000062.