


HIGHLIGHTS
1. Critical Discoveries and Outcomes
• Sialidosis type 1 is a common metabolic disorder that often draws the attention of ophthalmologists due to the presence of macular cherry-red spots. However, it is important to note that ocular findings such as lens opacities, nystagmus, and optic atrophy are significant diagnostic clues in affected patients.
2. Methodological Innovations
• For sialidosis type 1, ocular assessment should extend beyond structural imaging to include detailed functional testing. Early, comprehensive ophthalmic evaluation with multimodal imaging and functional assessment is essential for the timely diagnosis and management of type 1 sialidosis.
3. Prospective Applications and Future Directions
• The c.544A>G variant is the most frequent NEU1 mutation of sialidosis type 1 and may be associated with higher rates of lens opacities and visual evoked potential abnormalities.
Sialidosis is a rare lysosomal storage disorder. It is mainly characterized by a lack of the enzyme alpha-N-acetyl neuraminidase.[1-2] This autosomal recessive disease comes from mutations in the neuraminidase 1 (NEU1) gene, which is on chromosome 6p21.33. The NEU1 gene makes the enzyme alpha-N-acetyl neuraminidase (sialidase).[1,3] Sialidase starts the breakdown of glycoproteins and oligosaccharides or polysaccharides. It takes part in the breakdown of sialylated glycoconjugates within lysosomes.[3-4] The lack of this enzyme causes sialic acid-containing glycopeptides and oligosaccharides to build up inside cells. This disrupts normal cellular function and leads to a range of clinical symptoms.[3,5]
The disorder is generally divided into two types. Each type differs in severity and the age at which it starts. Type I sialidosis is also called the ‘normosomatic’ or cherry-red spot-myoclonus syndrome. It usually shows up later in life and is less severe than Type II.[6-8] Patients with this type often have symptoms like cerebellar ataxia, myoclonic epilepsy, nystagmus, and progressive visual loss, including blurred vision and color vision problems.[1,6] The clinical symptoms can range from almost no symptoms to more severe neurological and visual symptoms. Type II sialidosis appears earlier and is divided into congenital, infantile, and juvenile forms. Each form is marked by increasing severity and more abnormal physical features.[9]
The genetic situation of sialidosis has become clearer. About 40 different mutations in the NEU1 gene have been found. This shows the molecular basis of this complex disease.[2,10-11] Because these disorders are rare and the initial symptoms are not specific, often showing as a cherry-red spot in ophthalmology, they can be hard to detect and are often misdiagnosed.
Case presentation
Case report
This study describes a 7-year-old female patient. Written informed consent was obtained from the patient’s legal guardians. This study adhered to the Declaration of Helsinki and followed the Case Report (CARE) reporting guidelines.
The patient, a 7-year-old girl, first went to the Department of Ophthalmology at Lufeng People's Hospital in September 2023. She complained that her vision in both eyes had been getting worse for four months. Her visual acuity in both eyes was 0.16. At that time, her first fundus photography showed yellow-white lesions in the macular area of both eyes. And OCT showed that the superficial layers of the retina at the posterior pole were hyperreflective. But no clear diagnosis was made during this visit. Her guardians were told to take her to a higher-level ophthalmic center for further evaluation.
She had her first consultation at Zhongshan Ophthalmic Center in November 2023. According to the medical records, her main complaint at that time was that her vision in both eyes had been declining for six months. During this visit, her uncorrected visual acuity was 0.1 in the right eye and 0.16 in the left eye. Similar macular yellow-white lesions were seen again. The diagnosis was Stargardt disease in both eyes. She was told to come back for a follow-up if her symptoms got worse.
Over the next nine months, her binocular visual acuity got even worse. In July 2024, she went back to Zhongshan Ophthalmic Center for evaluation. During this visit, a detailed medical history was taken and a comprehensive ophthalmic examination was done. She was thin, had lost weight, had long limbs and fingers, had problems with her gait, and had ataxia. Her parents said she had foot pain at age 3, developed gait problems and unstable walking at age 4, and generally did not like to walk or run. Also, her stereoscopic vision acuity decreased. The parents said there were no consanguineous marriages or family history of similar conditions.
Her OCT showed beam-like high reflectivity in the superficial retinal layers of the central macular area, which matched the cherry-red spot regions. It also showed increased reflectivity in the ganglion cell layer of both eyes, and the boundaries between the ganglion cell and nerve fiber layers were not clear (Figure 1). FAF showed a ring of hyperautofluorescent regions around the fovea (Figure 2). Microperimetry suggested that the macular visual function in both eyes had declined, more severely in the right eye (Figure 3). Electroretinograms (ERG) showed that both eyes had normal responses for rods and cones. The visual evoked potential (VEP) showed significant visual pathway dysfunction. The right eye had severe abnormalities, with prolonged P100 latency, reduced amplitude, and no responses under both 60' and 15' stimuli. The left eye had similar but less severe abnormalities, including prolonged P100 latency, reduced amplitude under 60' stimulus, and no responses under 15' stimulus (Figure 4).
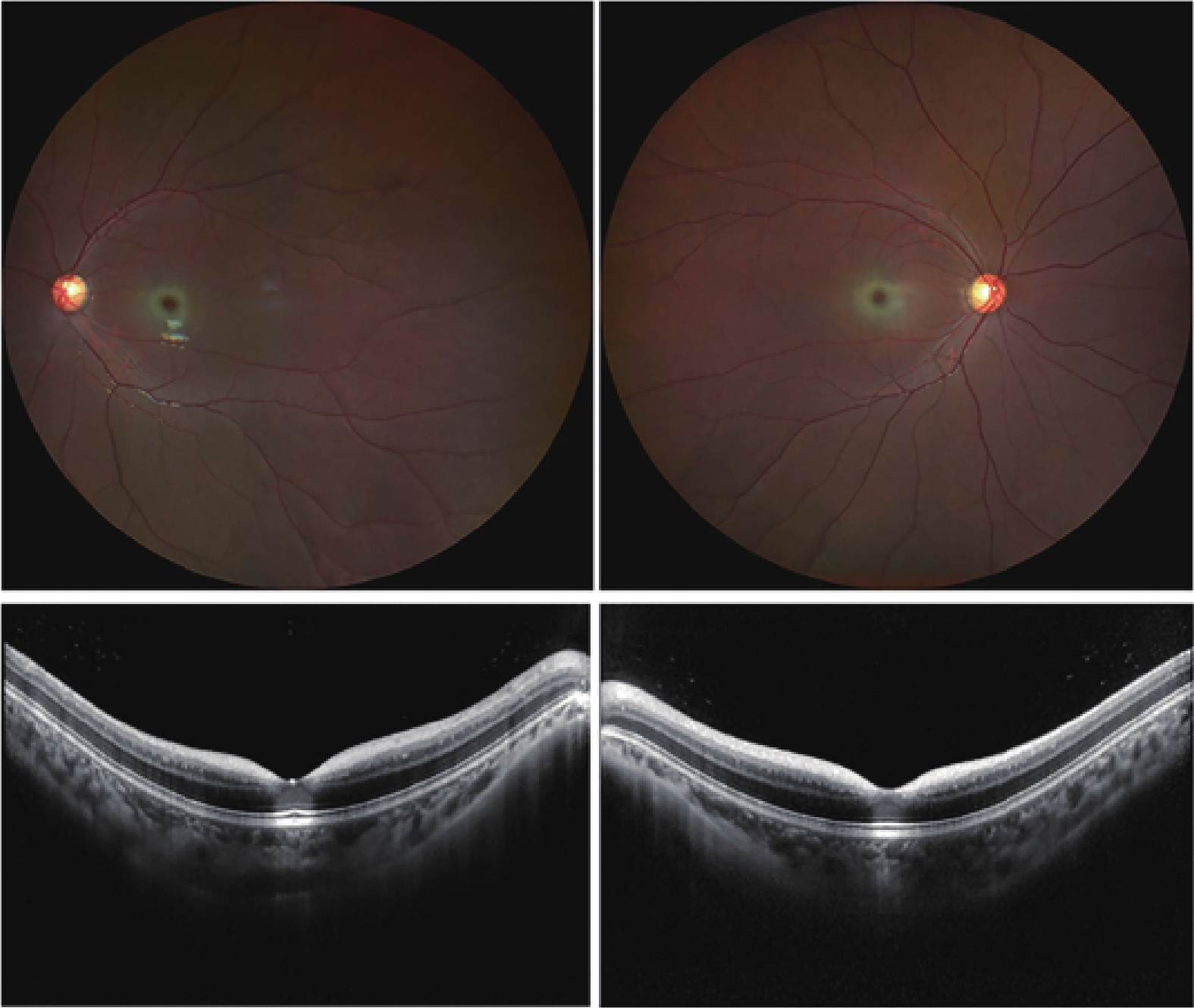
Fundus photography revealed bilateral macular cherry-red spots and temporal pallor of the optic discs in both eyes. Optical coherence tomography demonstrated beam-like high reflectivity in the superficial retinal layers corresponding to the cherry-red spot regions observed in fundus photography. Additionally, increased reflectivity was noted in the ganglion cell layer of both eyes, with indistinct boundaries between the ganglion cell and nerve fiber layers.

Fundus autofluorescence revealed hyperautofluorescent areas encircling the fovea.
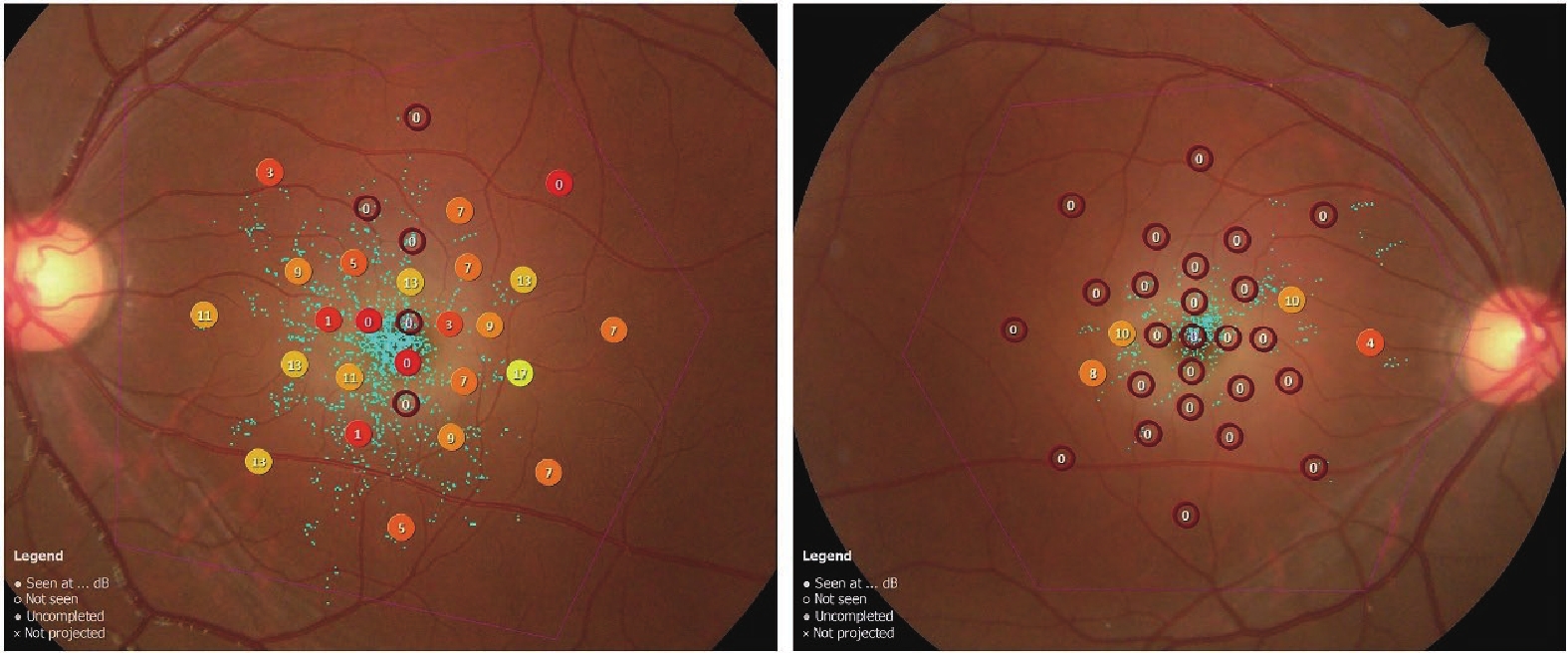
Microperimetry showed a decline in macular function in both eyes, more significantly in the right eye. The sensitivity values ranged from 0 to 34 decibels.
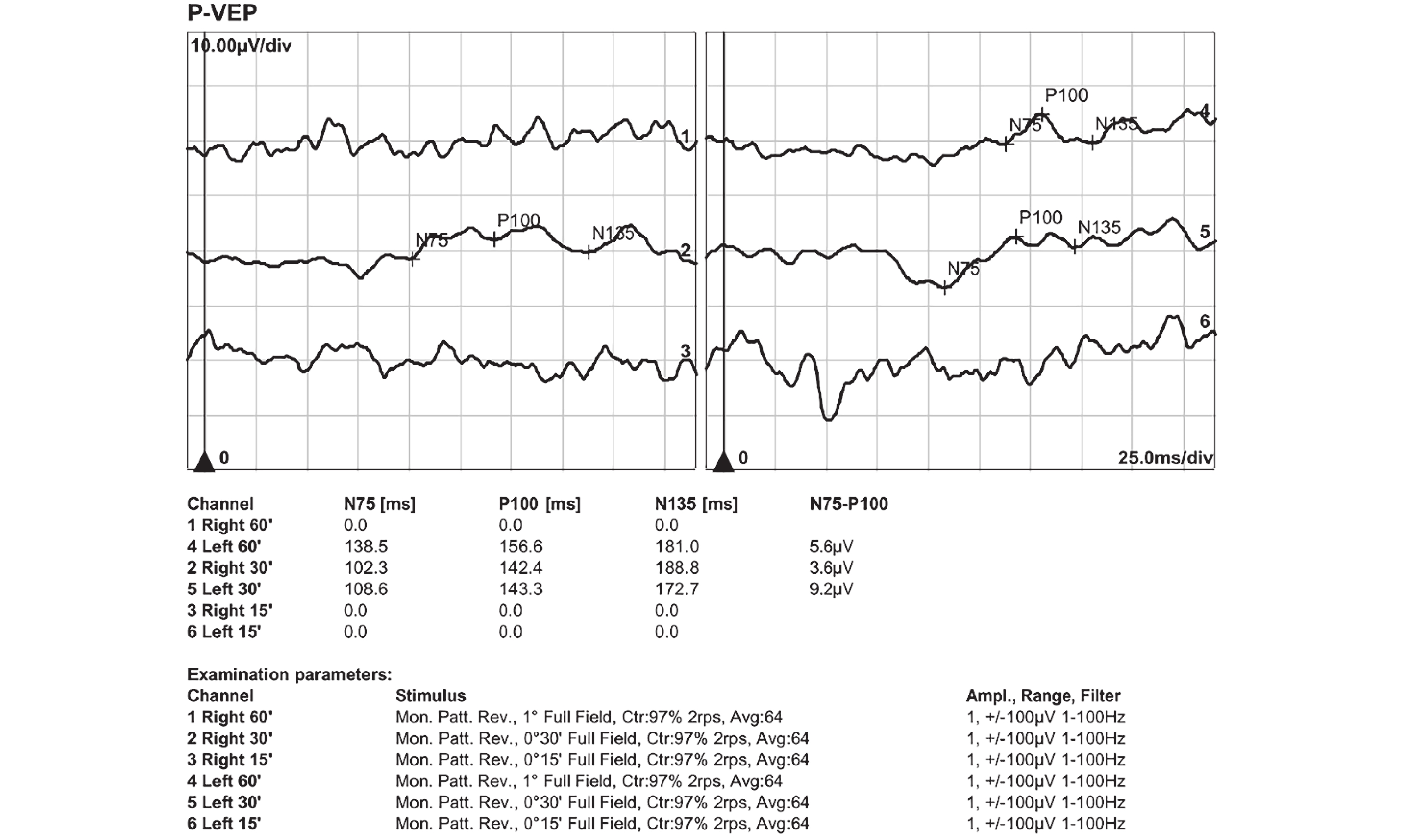
The visual evoked potential results revealed significant abnormalities in both eyes. For the right eye, under 30' stimulus, the P100 latency was prolonged to 142.4 ms (normal range: 95–115 ms), with a reduced amplitude of 3.6 µV (normal range: ≥5 µV). No responses were recorded under 60' and 15' stimuli, indicating severe dysfunction. For the left eye, the P100 latency under 30' and 60' stimuli was 143.3 ms and 156.6 ms, respectively, both markedly prolonged. The amplitudes were 9.2 µV under 30' stimulus, within the normal range, and 5.6 µV under 60' stimulus, at the lower limit of normal. No responses were recorded for the left eye under 15' stimulus.
Laboratory tests found two pathogenic mutations in the NEU1 gene associated with sialidosis (c.544A>G and c.239C>T), which she inherited from her father and mother respectively. Her younger brother had his mother's NEU1 (c.239C>T) mutation but had a normal clinical phenotype.
In the end, the patient was diagnosed with sialidosis type 1. It was suggested that her parents take her to the endocrinology and neurology departments of a comprehensive hospital for consultations. Since there is no treatment for sialidosis, she is currently being followed up by her ophthalmologist and neurologist.
Literature review
A comprehensive literature search was carried out in PubMed, Web of Science, and the China National Knowledge Infrastructure (CNKI) from when the databases started to June 30, 2025. The search terms were “sialidosis type 1”, “NEU1”, “cherry-red spot”, and “ocular manifestations”. The Inclusion criteria were as follows: (1) original studies, case reports, or case series where patients were diagnosed with sialidosis type 1 based on strict clinical evaluation and confirmed NEU1 gene mutations; (2) studies that described ocular clinical manifestations or ophthalmic examination findings; (3) individual-level clinical information was available; (4) publications were written in English or Chinese. The exclusion criteria were: (1) studies about patients with sialidosis type 2 instead of type 1; (2) cases labeled as sialidosis type 1 but without enough clinical or genetic evidence, resulting in an uncertain diagnosis; (3) studies that only described systemic manifestations without ophthalmic information; (4) incomplete case descriptions that only reported partial clinical findings without corresponding examination results. After removing duplicates and screening titles, abstracts, and full texts, a total of 35 studies were included. A PRISMA-style flow chart shows the literature selection process (Figure 5).
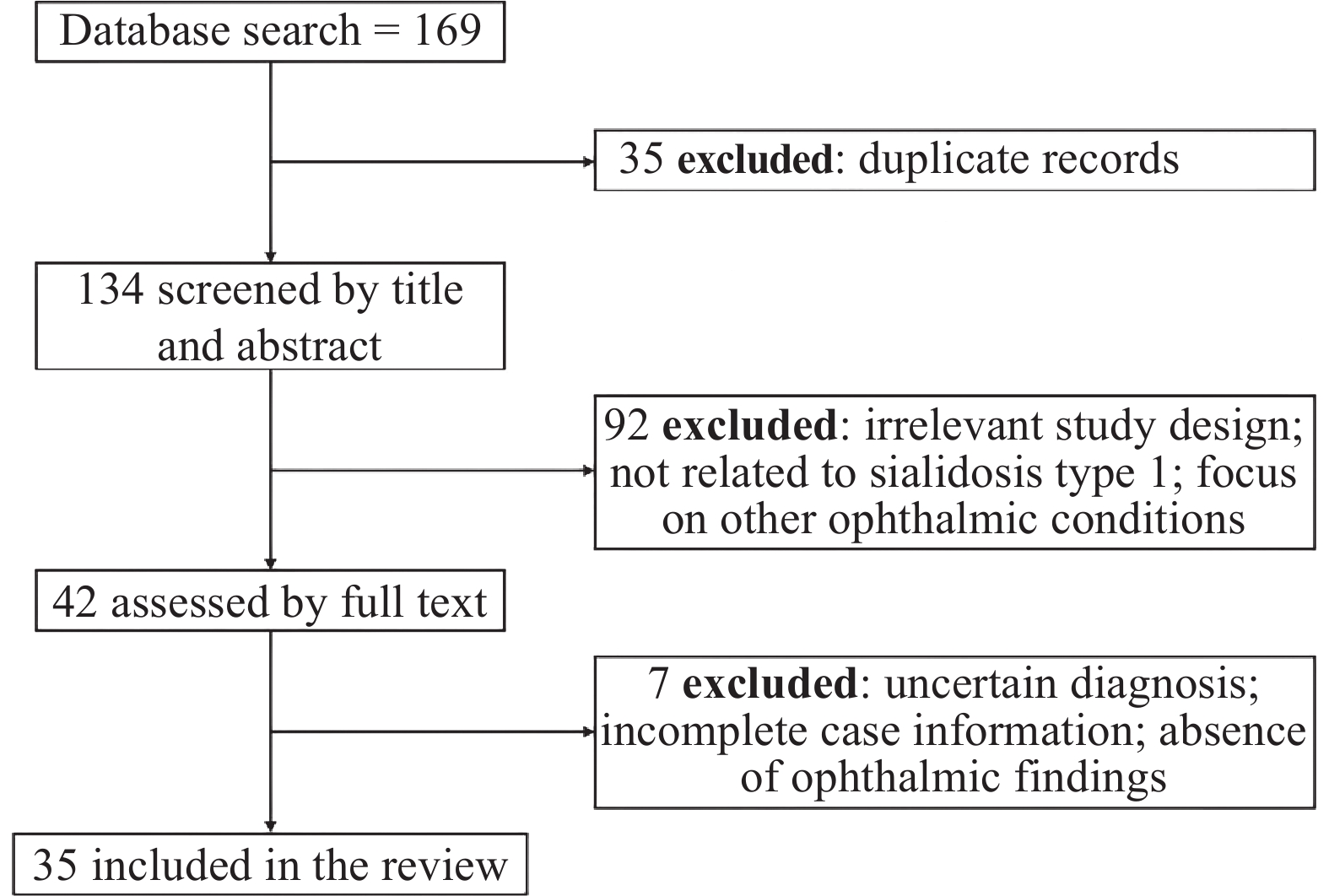
A total of 169 records were identified through database searches (PubMed, Web of Science, and CNKI). After removing 35 duplicate records, 134 articles were screened by title and abstract. Of these, 92 were excluded due to irrelevant study design, lack of relevance to sialidosis type 1, or focus on other ophthalmic conditions. The remaining 42 full-text articles were assessed for eligibility, and 7 were excluded because of uncertain diagnosis, incomplete case information, or absence of ophthalmic findings. Finally, 35 studies met the inclusion criteria and were included in the review.
Our literature review included 85 patients with sialidosis type 1 from previous studies and the current study, and we made a detailed summary (Supplementary Table 1). The combined results show that the average age of onset for patients reported in the literature is (16.3 ± 6.6) years, and the average age at examination is (28.2 ± 12.5) years. The gender ratio was 43 males to 42 females. We also summarized and analyzed the metabolic abnormalities reported in the literature. Our review shows that 70.6% of patients with sialidosis type 1 have metabolic abnormalities, mainly including elevated urinary sialic acid levels and reduced neuraminidase content and activity in fibroblasts. It is worth noting that many patients without documented metabolic abnormalities, including the present case, did not have specific tests for sialic acid or neuraminidase activity but were evaluated with routine metabolic tests such as blood tests. So, the actual proportion of metabolic abnormalities among these patients may be higher.
We did the first comprehensive and detailed review of the ocular manifestations and examination findings in patients with sialidosis type 1 (Supplementary Table 2). Our analysis shows that 27.1% of patients have ocular symptoms as their first manifestation and are first diagnosed in ophthalmology clinics. Visual impairment is the most common ocular symptom, affecting 58.0% of patients. The characteristic feature, macular cherry-red spots, is seen in 57.0% of cases. Besides cherry-red spots, we found three other common ocular findings: lens opacities (62.1%), nystagmus (38.2%), and optic atrophy (32.0%). Notably, the prevalence of lens opacities is higher than that of cherry-red spots, making it one of the most underrecognized yet highly frequent ocular signs in patients with sialidosis type 1.
For eye examination results, 76.7% of patients with sialidosis type 1 show OCT abnormalities. These are mainly hyperreflectivity in the macular inner layer. 66.7% of patients have visual field defects, usually localized or in specific quadrants. All five patients who had FAF tests showed hyperautofluorescent areas around the fovea. But only 3 of 11 patients (27.3%) who had ERG tests showed abnormalities, specifically cone dysfunction. VEP abnormalities are more common, affecting 78.1% of patients. These mainly show as prolonged VEP latency.
We also summarized and analyzed the NEU1 gene mutations in sialidosis Type 1 in Supplementary Table 3. Our findings show the most common NEU1 gene mutation is homozygous c.544 A > G, found in 32.9% of cases. The next most common is compound heterozygous c.544A > G; c.239C > T, found in 11.8%. Supplementary Table 3 shows the c.544 A > G variant is very common. The mutation in our case, c.544A > G; c.239C > T, is one of the commonly reported types. Supplementary Figure 1 shows NEU1 protein changes in sialidosis Type 1.
Our analysis of patients with the c.544A > G mutation found more lens opacities (75.0%, 6 of 8 patients) and much more VEP abnormalities (96.8%, 30 of 31 patients).
Discussion
We reported a case of a 7-year-old girl who presented to Zhongshan Ophthalmic Center with bilateral visual impairment. Slit-lamp examination showed bilateral cataracts. Fundus examination found characteristic macular cherry-red spots and temporal pallor of the optic discs in both eyes. Besides ocular symptoms, the patient had many systemic pathological changes. So we suspected a diagnosis of inherited retinal disorder with systemic involvement. We suggested further genetic tests and a comprehensive ophthalmological evaluation, including visual function assessment, to confirm the diagnosis of sialidosis. It should be noted that the patient had been misdiagnosed with Stargardt disease before. Our case is different from similar reports. We did not just rely on genetic tests, also did thorough retinal structural and functional assessments to help with the diagnosis.[12]
Macular cherry-red spots are often linked to central retinal artery occlusion and other ophthalmic diseases.[13] At the first evaluation, we considered several differential diagnoses for the patient's ocular symptoms, like Stargardt disease, Best disease, and central retinal artery occlusion. But Stargardt disease usually shows scattered yellow dots, not a confluent yellow-white area as in our patient. And the patient's retinal thickness was normal in both eyes, with no sign of macular atrophy.[14] Best disease is a hereditary macular degeneration. It shows egg-yolk-like lesions in the macula. But these lesions are usually oval, unlike the confluent yellow-white area in our patient. And the OCT findings did not support this diagnosis.[15] Central retinal artery occlusion (CRAO) was also unlikely, because the visual loss progressed slowly and there was no retinal edema.[16] Finally, we considered the patient's systemic symptoms and suspected a hereditary disease with ocular and systemic involvement. So we suggested genetic testing, which confirmed the diagnosis.
Although both CRAO and sialidosis type 1 can cause macular cherry-red spots, their underlying mechanisms are very different. In CRAO, the cherry-red spot appears because of retinal ischemia caused by central retinal artery obstruction. This ischemia makes the ischemic retina swell and become opaque, except for the fovea. There are no ganglion cells in the fovea, and the thinner retinal layers let the underlying red choroid show through, creating the cherry-red look.[13] In contrast, in sialidosis type 1, the cherry-red spot is due to the buildup of metabolic material in the retinal ganglion cells, especially in the macular region where these cells are densely packed. Since the fovea has no ganglion cells, it stays transparent, which is a sharp contrast to the surrounding opaque retina.[17]
OCT imaging shows unique features of sialidosis. For example, there is beam-like hyperreflectivity in the central macula and ganglion cell layer. This is because of the high ganglion cell density and the deposition of metabolic byproducts, which make the boundary between the ganglion cell layer and the nerve fiber layer.[1,3,12,18-19] In this case, further quadrant analysis of the retinal nerve fiber layer (RNFL) using OCT showed a significant thinning of the superior and inferior RNFL in both eyes. Interestingly, this was also seen in the study by Sobral et al. This suggests that sialic acid deposition may not only affect the ganglion cell layer but also contribute to damage in the RNFL.[17]
Also, macular cherry-red spots are a typical feature of sialidosis and other lipid/lysosomal storage disorders, like sphingolipidoses, gangliosidoses, Fabry disease, Niemann-Pick disease, etc.[1,13,17,19-22] These disorders are characterized by the buildup of complex lipids in the retina. This results in grayish, peri-macular deposits. The fovea is not affected but looks unusually red and stands out against the surrounding infiltrates.[13] The presence of systemic pathological changes in our patient and the macular cherry-red spots helped with the differential diagnosis of central nervous system storage diseases and other ophthalmic conditions, such as central retinal artery occlusion, Best disease, and traumatic retinal edema.
At present, treatment options for sialidosis type 1 are limited. Available therapies mainly focus on symptom management, like antiepileptic drugs and medications to control myoclonus.[23-24] But there is no evidence that these treatments directly affect ocular manifestations, including macular cherry-red spots. As ophthalmologists, we should consider whether therapeutic interventions can improve ocular signs. This emphasizes the potential role of combined local treatments along with systemic management. Regular follow-up and careful monitoring of ocular changes should be a priority. Looking forward, advancements in targeted therapies, like gene therapy, may address the underlying pathophysiology of sialidosis and improve both systemic and ocular outcomes.[25-26]
Besides this case, we did a systematic review of ocular manifestations in sialidosis type 1. Besides the characteristic macular cherry-red spot, lens opacities, nystagmus, and optic atrophy were often reported. Lens opacities were a common but underrecognized feature. These findings suggest that ocular involvement in sialidosis type 1 is broader than traditionally thought and needs more attention in clinical practice.
For ophthalmic examinations, OCT and VEP abnormalities were often seen and seemed to be more sensitive in detecting disease-related changes than ERG, which was often normal. FAF consistently showed pericentral hyperautofluorescence in the limited number of reported cases. This indicates its potential diagnostic value, but larger studies are needed to confirm it.
We further analyzed genotype–phenotype associations. We found that the c.544A>G mutation, which is common in Asian populations,[1,3,5,19,22,27-34] was associated with a higher frequency of lens opacities and VEP abnormalities. This suggests a possible predisposition to lens metabolic disturbance and optic nerve involvement.
Multimodal ophthalmic assessment is important in distinguishing sialidosis-related macular changes from other causes of cherry-red spots.[22] Characteristic inner retinal hyperreflectivity on OCT and pericentral hyperautofluorescence on FAF, along with functional deficits detected by microperimetry and VEP, provide valuable diagnostic clues that are not usually seen in other conditions.
It should be noted that nearly one-quarter of patients first showed ocular symptoms and were first evaluated in ophthalmology clinics. But many reports put too much emphasis on the presence of cherry-red spots and did not pay enough attention to other important ocular features. This shows the need for comprehensive ophthalmic evaluation and more awareness among ophthalmologists.
Conclusions
We know that this study is the first to report sialidosis type 1 with a comprehensive ophthalmic and genetic assessment. It is also the first systematic review that focuses on its ocular manifestations. But the disease is rare, and the published data are heterogeneous. This limits the generalizability of the findings. It shows that there is a need for future multicenter studies with standardized ophthalmic assessments.
In summary, sialidosis type 1 may first show ocular symptoms and has a wide range of ophthalmic involvement. An early and comprehensive ophthalmic evaluation, which goes beyond identifying cherry-red spots and includes checking the lens status, assessing the optic nerve, and doing functional testing, may help with timely diagnosis and management.
List of abbreviations
CRAO, central retinal artery occlusion
ERG, electroretinography
FAF, fundus autofluorescence
RNFL, retinal nerve fiber layer
NEU1, neuraminidase 1
OCT, optical coherence tomography
VEP, visual evoked potential
Correction notice
None
Acknowledgments
We gratefully acknowledge the support of the Guangdong Basic Research Center of Excellence for Major Blinding Eye Diseases Prevention and Treatment.
Author Contributions
(I) Conception and design: Xiufeng Zhong
(II) Administrative support: Xiufeng Zhong
(III) Provision of study materials or patients: Xiufeng Zhong
(IV) Collection and assembly of data: Xiufeng Zhong, Deming Wang
(V) Data analysis and interpretation: Deming Wang, Wenjing Yin
(VI) Manuscript writing: All authors
(VII) Final approval of manuscript: All authors
Conflict of Interests
None of the authors has any conflicts of interest to disclose. All authors have declared in the completed the ICMJE uniform disclosure form.
Patient consent for publication
Written informed consent for publication of this case report was obtained from the patient’s legal guardian.
Ethical Statement
This study was approved by the Ethics Committee of Zhongshan Ophthalmic Center. Written informedconsent to participate was obtained from the patient’slegal guardian.
Data availability statement
None
Open access
This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license).